The relative importance of each of the two mechanisms represents a controversial, unanswered question at the core of the C9-ALS/FTD field. Indeed, since the discovery C9orf72-linked ALS/FTD, many conflicting results have been published regarding the relative importance of each of the two mechanisms. The lack of a clear correlation between repeat length, for example, appears to argue against the toxic gain-of-function as being a major driver of disease pathology. In line with this a recent phase I clinical trial (BIIB078) of an antisense oligonucleotide (ASO), targeting G4C2 repeat RNA and thus also DPR generation, failed to show any clinical benefits. It is unclear, however, whether the ASO administration indeed significantly lowered the levels of repeat RNAs and DPRs. Arguing instead against the loss-of-function hypothesis, patients homozygous for the
C9orf72 repeat expansion, and thus expressing less
C9orf72, do not show a more serve phenotype than those with
C9orf72 haploinsufficiency [
11]. In addition,
C9orf72 knockout mice do not show any ALS/FTD-associated neurodegenerative phenotypes [
12]. Although this remains an important unanswered question to be addressed in the field, the two mechanisms likely act in coordination, and it is therefore important to continue investigating both hypotheses.
Interestingly, the overexpression of a sufficient number of G4C2 repeats per se (i.e., without modulation of
C9orf72 gene dosage) in vitro [
13,
14,
15] as well as in mice [
16,
17], zebrafish [
18,
19], and
Drosophila melanogaster [
20,
21] is sufficient to induce toxicity and neurodegeneration. These can be bidirectionally transcribed into repeat RNAs that subsequently aggregate, a general hallmark of non-coding repeat expansion diseases. Indeed, samples from patients with C9-ALS/FTD and patient-derived induced pluripotent stem cells (iPSCs) have been shown to contain nuclear, as well as some cytoplasmic, foci of aggregated RNA [
18,
20,
22,
23,
24]. Further, these sense and anti-sense repeat RNAs can undergo non-canonical, repeat-associated non-AUG (RAN) translation to produce five distinct dipeptide repeats (DPRs): poly-PA, poly-PR, poly-GA, poly-GP, and poly-GR [
25,
26,
27,
28,
29].
2. The Impact of C9-Associated Toxic Repeats on Protein Degradation Pathways
A key neuropathological hallmark in C9-ALS/FTD is the formation of star-shaped cytoplasmic DPR inclusions which are ubiquitin- and p62-positive [
30,
31], indicative of an involvement of the ubiquitin–proteasome system (UPS) and of macroautophagy (hereafter autophagy) in the targeted degradation of DPRs. Consistent with this, the pharmacological inhibition of either pathway leads to an increased accumulation and aggregation of distinct DPRs in cell culture [
14,
32]. However, several recent studies suggest that, while DPRs are targets of UPS- and autophagy-mediated clearance, they might also play a major role in disrupting both pathways (
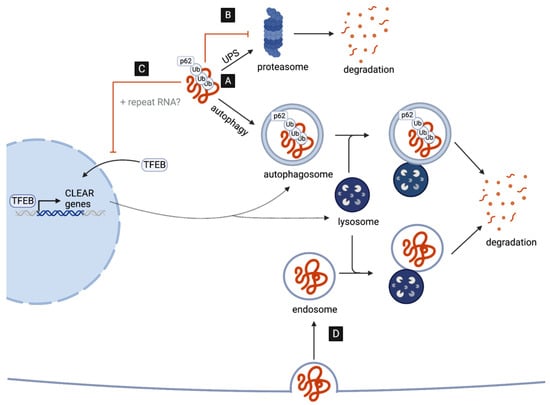
Figure 1), as discussed in detail in the next two chapters.
Figure 1. DPRs as targets and disruptors of cellular clearance pathways. (A) C9-associated DPRs are targeted for degradation by the ubiquitin–proteasome system (UPS) and the autophagy pathway. (B) DPRs impair the UPS by directly interacting with and inhibiting the 26S proteasome. (C) DPRs (and repeat RNAs) disrupt the autophagy pathway reducing the nuclear translocation of TFEB, the master transcriptional regulator of autophagy genes (CLEAR network). (D) G4C2-induced impairment in lysosome function may aid the cell-to-cell transmission of DPRs via endocytosis.
2.1. Poly-GA Inhibits Protein Degradation via the UPS
The UPS is the major cellular pathway responsible for the degradation and recycling of short-lived, soluble proteins, and indeed appears to be important for DPR degradation [
14,
32]. In addition to being possible targets of UPS-mediated degradation, DPRs in fact appear to inhibit UPS activity [
13,
14,
15,
33,
34,
35,
36,
37]. Interestingly, several mutations in genes involved in the UPS have been associated with both ALS and FTD [
4], highlighting the UPS as a common point of vulnerability in both neurodegenerative diseases. While all DPRs, with the notable exception of poly-PA, have been associated with disrupted UPS, the underlying mechanisms remain unclear.
Several studies have shown that UPS factors co-localize with cytoplasmic poly-GA inclusions in cell culture [
13,
14,
15,
33,
34], in animal models, and in the brains of patients with C9-ALS/FTD [
35]. Pointing to a more direct impairment of the UPS system by accumulation poly-GA, it has recently been shown that the 26S proteasome, which is ultimately required to degrade ubiquitin-tagged proteasomal substrates, is sequestered into poly-GA aggregates in cultured neurons [
33]. In situ structural analysis of neuronal poly-GA aggregates suggests that these may force the sequestered proteasomes to become stuck in a highly transient intermediate state, which is usually associated with substrate translocation. This may lead to stalled degradation of ubiquitinated substrates, which could explain observed reductions in proteasome activity [
33].
The specific consequences of Impaired proteasome function in the context of C9-linked ALS/FTD remains to be further investigated. However, independent of C9-ALS/FTD, the inhibition of the 26S proteasome has also been shown to lead to the cytoplasmic mislocalization and aggregation of TAR DNA-binding protein 43 (TDP43) [
38,
39], a second aggregation-prone protein associated with the vast majority of familiar and sporadic forms of ALS. Indeed, some TDP-43 pathology has been observed in patients with C9-ALS/FTD [
28,
40,
41], although it appears to occur after DPR pathology [
42,
43]. Whether proteasomal inhibition by poly-GA may be a contributing factor to TDP-43 pathology therefore warrants further investigation. Indeed, primitive evidence suggests that poly-GA aggregates are able to induce TDP-43 mislocalization in cell culture, and that this mislocalization is dependent on GA-induced inhibition of the proteasome [
34]. Interestingly, proteasome inhibition was shown to occur in a cell-autonomous and non-cell-autonomous manner. Specifically, cytoplasmic TDP-43 was observed in rat primary hippocampal neurons cultured with cell supernatant from GA-transduced cells. Such TDP-43 mislocalization was eliminated when depleting the culture media of poly-GA with anti-GA antibodies. While is remains to be determined whether this is through cell-to-cell transmission, this mechanism may help to explain why DPR inclusions and TDP-43 pathology predominantly occur in distinct neurons within the brains of patients with C9-ALS/FTD [
41].
2.2. C9-Associated Toxic Repeats Disrupt Autophagosome and Lysosome Biogenesis
In addition to the UPS, the autophagy pathway is another essential contributor to intracellular protein clearance, which predominantly targets insoluble, aggregated, and long-lived proteins. The autophagy pathway involves the recognition of ubiquitin-tagged proteins destined to clearance by adaptor proteins, such as p62/SQSTM1, promoting engulfment by the forming autophagosome, a specialized double-membrane organelle [
44]. In neurons, autophagosomes are predominantly formed in distal regions of the axon and, as they undergo retrograde transport toward the soma, they mature and ultimately fuse with lysosomes, a process critical for neuronal longevity [
45]. Importantly, lysosomes contain digestive enzymes, activated by a low pH, which eventually break down the autophagic cargo, allowing the degraded products to be recycled back to the cell [
46].
Emerging evidence suggests that both repeat RNAs and DPRs may disrupt multiple steps of the autophagy pathway. The overexpression of G4C2 repeats in
Drosophila motor neurons, for example, leads to an accumulation of
Drosophila p62, accompanied by a reduction in the number of autophagosomal vesicles in both the soma and distal axons in vivo [
47,
48]. These data suggest that autophagy initiation, and specifically autophagosome formation, may be impaired. In line with this, G4C2 repeat expression results in reductions in the levels of mature autophagosomes in cell culture [
49].
How could autophagy induction be affected by the expression of G4C2 repeats? One possibility is that reductions in autophagosome formation may be, at least in part, a result of disruptions in TFEB, the master transcriptional regulator of autophagy and lysosomal biogenesis (
Figure 1C). Specifically, G4C2 toxicity appears to inhibit the nuclear import of TFEB in
Drosophila as well as in cell culture [
47,
49]. Remarkably, dysfunction in nucleo-cytoplasmic transport and the nuclear pore complex (NPC) is emerging as a key contributor to disease pathogenesis [
50]. This includes disruptions induced by repeat RNAs in
Drosophila neurons and patient-derived iPSCs [
51,
52], as well as by DPRs. In fact, using
Xenopus laevis oocytes as a model system, poly-PR has specifically been shown to bind to the central channel nuclear pore to induce a block in the transport of macromolecules between the nucleus and cytoplasm [
53]. Additionally, both cytoplasmic poly-GA and poly-GR aggregates appear to sequester components of the NPC in the brains of DPR-expressing mice and patients with C9-ALS/FTD [
35,
54]. This includes the nucleoporin POM121, which when overexpressed leads to a rescue of TFEB nuclear localization and autophagy initiation in cell culture [
49].
In addition to promoting autophagosome formation, TFEB is essential in mammals and in flies for the expression of genes required for lysosome biogenesis and function [
55,
56,
57]. Thus, it is also possible that lysosomes might be defective in the presence of repeat RNAs and DPRs. Consistent with this possibility, G4C2 overexpression results in reduced cleavage and activation of a
Drosophila cathepsin and might result in reduced lysosome acidification. Indeed, the overexpression of vacuolar ATPase (V-ATPase) genes, encoding the main proton pump required for lysosome acidification, appear to suppress G4C2-induced neurodegeneration in
Drosophila [
47]. Interestingly, the specific overexpression of only poly-GA in human cells has been shown to lead to the accumulation of mature autophagosomes, as well as p62 and ubiquitin, which is indicative of lysosomal impairment but not necessarily disruptions in autophagosome formation [
58]. This discrepancy may be a result of the different system used, as well as the differences arising from the expression of a single DPR versus all DPRs simultaneously (e.g., G4C2 repeat overexpression).
Despite the accumulating evidence of the G4C2-induced impairment of autophagy and lysosomal biogenesis, it is important to note that
C9orf72-deficient cells also display reduced levels of autophagy initiation and impaired lysosome biogenesis, leading to enhanced DPR accumulation and increased neurotoxicity, indicating that the product of
C9orf72 might play a role in physiologic activation of autophagy [
59,
60,
61,
62,
63,
64,
65]. This is not surprising as C9orf72 is likely to act as an GDP/GTP exchange factor (GEF) for a number of RabGTPases regulating the early steps of autophagy and endocytic trafficking [
66].
While reduced autophagy may thus represent an important contributor to C9-ALS/FTD pathogenesis, the consequences of reduced autophagy might extend beyond the impaired degradation of the DPRs themselves. Intriguingly, Marchi et al. recently proposed that reduced lysosome acidification may represent a mechanism by which endocytosed poly-GA aggregates can circumvent lysosomal degradation, thus enabling the retention and spread of poly-GA aggregates via the endocytic–exosomal pathway [
67]. Although the mechanisms of cell-to-cell transmission may vary, all DPRs have indeed been observed to spread between cells in vitro [
34,
68,
69,
70]. Thus, it will be interesting to study (in the future) whether the endocytic–exosomal pathway represents a predominant mechanism of cell-to-cell transmission in C9-ALS/FTD (
Figure 1D).