Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Qingming Fang | -- | 3630 | 2024-01-19 22:43:36 | | | |
| 2 | Lindsay Dong | Meta information modification | 3630 | 2024-01-22 02:34:03 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Fang, Q. Versatile Attributes of MGMT. Encyclopedia. Available online: https://encyclopedia.pub/entry/54137 (accessed on 27 July 2026).
Fang Q. Versatile Attributes of MGMT. Encyclopedia. Available at: https://encyclopedia.pub/entry/54137. Accessed July 27, 2026.
Fang, Qingming. "Versatile Attributes of MGMT" Encyclopedia, https://encyclopedia.pub/entry/54137 (accessed July 27, 2026).
Fang, Q. (2024, January 19). Versatile Attributes of MGMT. In Encyclopedia. https://encyclopedia.pub/entry/54137
Fang, Qingming. "Versatile Attributes of MGMT." Encyclopedia. Web. 19 January, 2024.
Copy Citation
O6-methylguanine-DNA methyltransferase (MGMT or AGT) is a DNA repair protein with the capability to remove alkyl groups from O6-AlkylG adducts. Moreover, MGMT plays a crucial role in repairing DNA damage induced by methylating agents like temozolomide and chloroethylating agents such as carmustine, and thereby contributes to chemotherapeutic resistance when these agents are used.
MGMT
AGT
base excision repair (BER)
mismatch repair (MMR)
direct reversal
homologous recombination (HR)
1. Introduction
Chemical insults, both endogenous (e.g., S-adenosylmethionine) and exogenous (e.g., alkylating agents), constantly attack cellular DNA, resulting in DNA lesions [1][2][3][4]. Among these chemicals, alkylating agents play a significant role in causing DNA damage [5]. These agents are widely present in the environment, used as anticancer compounds in clinical settings [1][2][3][4][6][7][8][9][10], and can be endogenously generated during normal cellular processes. For example, S-adenosylmethionine, a methyl donor involved in various cellular reactions, has been shown to induce DNA methylation damage [3][11][12]. The attack of alkylating agents on DNA can lead to different types of lesions on the heterocyclic bases or backbone [3][5][13][14][15][16]. Most of these resulting adducts are mutagenic or toxic, prompting cells to develop various proteins for their detection and repair [13][17]. Interestingly, many of these alkylation lesions are repaired through direct removal of the adduct [5]. O6-alkylguanine-DNA alkyltransferase or O6-methylguanine-DNA methyltransferase, known as AGT or MGMT, is an enzyme responsible for the repair of alkylated DNA through a process known as “suicide” repair [3][4][13]. MGMT expression is primarily regulated through epigenetic modifications [18][19][20], and extensive research has indicated that reduced MGMT expression is attributed to the methylation of the CpG island that spans 762 base pairs and contains 98 CpG sites found in the MGMT promoter region [21][22][23][24]. Mechanistically, the primary function of MGMT involves the transfer of the methyl group located at the O6 site of guanine to cysteine residues located within the MGMT active site, thereby preventing mutations, cell death, and the development of tumors caused by alkylating agents [3][13].
2. Repair Mechanism
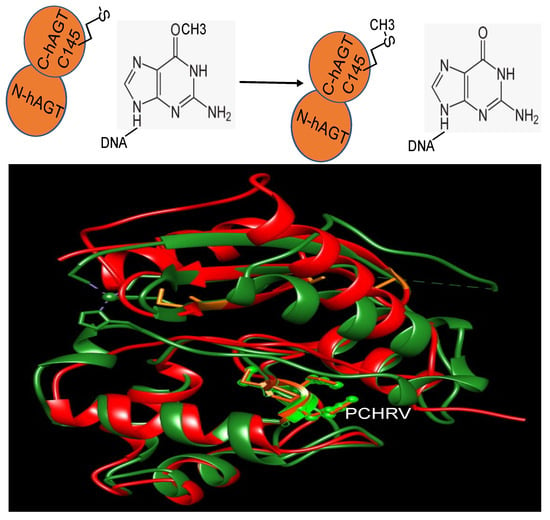
The initial fully characterized MGMT or AGT was the Ada gene product from E. coli [25][26]. It performs the repair of O6-methylguanine (m6G) in DNA through the direct transfer of the methyl group from the DNA to a cysteine residue within the protein itself (Figure 1) [25][26]. The inducibility of the Ada protein in response to alkylation damage facilitated its biochemical examination, enabling purification in substantial quantities after such treatment [27][28]. Subsequent investigations unveiled another MGMT in E. coli, Ogt [29][30].
Figure 1. The reaction of MGMT (top panel) and the structure overlay of Ada and hMGMT (bottom panel). Ada: red; hMGMT: green; the side chains of PCHRV are shown. The overlay is to show the structural similarity of both proteins.
MGMT is widely present in almost all organisms. It is easily identifiable due to the characteristic protein sequence surrounding the central Cys site that acts as the acceptor for alkyl groups: (I/V)PCHR(V/I)-(I/V) (Figure 1) [13]. Multiple structures of hMGMT have been elucidated, providing valuable insights into its functioning [31][32][33][34]. The structure of MGMT comprises two domains. The C-terminal domain encompasses the active site pocket and DNA binding region, interconnected by a hinge formed by the conserved residue Asn137 (amino acid numbers provided refer to hMGMT) (Figure 1) [31][32][33][34]. The N-terminal domain plays a vital role in maintaining structural stability, and can stabilize the complex formed when the two domains are expressed separately [35]. The initial published structure revealed a similar fold to the C-terminal domain of Ada (C-Ada: One of MGMTs in E. coli), even in the N-terminal domain, despite a lower sequence similarity between the two proteins [31](Figure 1).
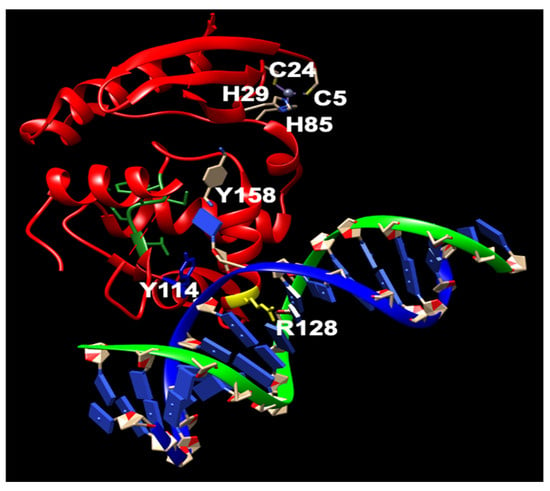
Figure 2. The structure of human MGMT repairing (modified from PDB ID: 1EH7). Y114: blue; R128: yellow; PCHRV: green; hMGMT: red; dsDNA: green and yellow. Nucleotides: blue. The key residues involved in the repair are shown.
Further biochemical investigations have revealed that although the zinc(II) site in MGMT is located away from the reaction center, its binding to a zinc(II) ion effectively lowers the pKa of Cys145 compared to the apo protein, thereby enhancing the reactivity of the protein [36]. hMGMT utilizes a helix-turn-helix motif to interact with DNA, but exhibits an unusual behavior of specifically targeting the minor groove [33]. The recognition helix, characterized by hydrophobic residues, tightly fits into the minor groove with minimal sequence-specificity [33]. The structural findings have led to the proposal of a comprehensive mechanism for the activation and repair of Cys145. This mechanism involves a hydrogen bonding network that activates the Cys145 residue, with a crucial Tyr residue (Tyr114) playing a role in identifying the damaged guanine, and providing a proton to facilitate the removal of the alkyl adduct.
3. The Function of N-Ada and the Potential Role of N-hMGMT
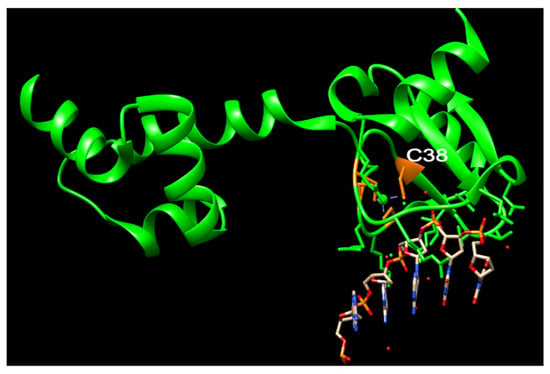
The Ada protein consists of two domains: a 20 kDa N-terminal domain (N-Ada) and a 19 kDa C-terminal domain (C-Ada) containing 354 amino acids (Figure 1) [37][38]. In E. coli, the Ada protein serves multiple functions. N-Ada is responsible for repairing the Sp-configurated methylphosphotriester, while C-Ada repairs the highly mutagenic m6G and m4T [37][38][39]. Additionally, Ada acts as a transcriptional activator that induces the robust expression of the ada, alkA, alkB, and aidB genes, thereby regulating the adaptive response to methylation resistance [40]. This response occurs after transferring the methyl group from the Sp-methylphosphotriester to the Cys38 residue [39]. The methylated N-Ada transforms into a potent DNA binder, enabling it to recognize the promoter regions of the Ada regulon and recruit RNA polymerase to initiate the transcription of the aforementioned four genes [17][39][41][42][43]. This specific Cys38 residue demonstrates selective activation, for it is the only Cys ligand that is not involved in any hydrogen-bonding interactions, and exhibits higher nucleophilicity compared to other Cys residues (Figure 3) [39][44][45][46].
Figure 3. The structure of N-Ada and its interaction with DNA chain (modified from PDB ID: 1U8B). N-Ada: green; zinc cluster with side chains of C38, C42, C69, and C72: orange; phosphodiesterase chains: red and orange. Nucleotides: blue and grey.
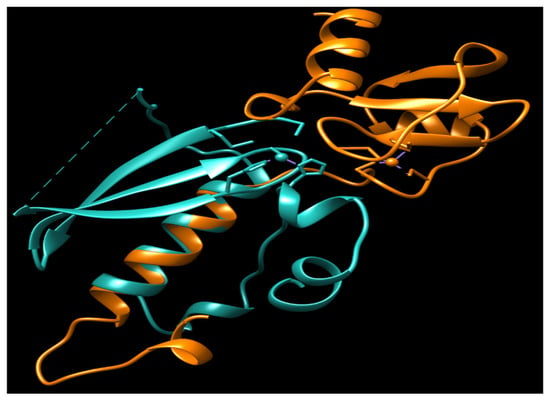
urrently, no N-Ada homologues have been identified in humans, and there is no evidence to suggest that methylphosphotriesters are repaired in human cells. The adaptive response to the methylation challenge is not clearly observed in eukaryotes. All the residues identified as being associated with the activity and DNA binding of hMGMT are located in the C-terminal domain (Figure 2) [32][33][47]. Structural and biochemical studies of hMGMT indicate that N-hMGMT may primarily serve a passive structural role, rather than directly participating in the protein’s functional activity [31][32][33][34]. Previous biochemical studies indicated that hMGMT exhibits more similarity to Ogt than to Ada [48][49]. However, the published structures of hMGMT have revealed a similar fold to Ada [31][32][33][34] (Figure 1 and Figure 4).
Figure 4. The overlay of N-Ada and N-hMGMT. N-Ada: orange; N-hMGMT: light sea green; Zn2+: small ball. Both N-Ada and N-hMGMT contain zinc binding sites and these sites locate in the surface of protein.
4. The Fate of Alkylated MGMT
Early reports suggested that the addition of an alkyl group to the Cys145 of hMGMT causes structural alterations, leading to its recognition by ubiquitin ligases and subsequent degradation by the proteasome [50][51][52]. This degradation process may be necessary for continuous repair, as the presence of alkylated MGMT or inactive mutants like C145A can interfere with the repair function of active MGMT molecules [53]. It has been proposed that the disruption of a salt bridge between Asn137 and the carbonyl oxygen of Met134, caused by a steric clash with the newly formed S-alkylcysteine, serves as the signal for this degradation [32]. These alterations destabilize the protein but have minimal impact on DNA binding [54][55].
Accumulating evidence suggests that alkylated MGMT functions as a transcriptional regulator, regulating the DNA damage response. hMGMT is found at active transcription sites, facilitated by interacting with RNA polymerase II, and it exhibits a preference for repairing m6G damage specifically in the transcription strands [56]. Upon alkylation, MGMT undergoes a conformational change that exposes VLWKLLKVV residues (codons 98 to 106), allowing it to bind to the estrogen receptor (ER), a critical transcription factor involved in regulating cell proliferation [57]. The binding of alkylated MGMT to ER prevents its interaction with steroid receptor coactivator-1, thereby inhibiting the activation of ER-regulated gene expression [57]. This suggests that alkylated MGMT functions as a transcription regulator, leading to cell cycle arrest. Additionally, alkylated MGMT suppresses the expression of deubiquitinating enzyme 3 (DUB3), thereby impacting chemoresistance in ovarian cancer [58]. It is currently unknown whether this effect is associated with alkylated N-hMGMT or alkylated hMGMT-C. Further research is necessary to elucidate this relationship. Previous studies have indicated that the expression of hMGMT can be modulated by glucocorticoids and partial hepatectomy [59][60].
5. The Role of hMGMT in Cancer Prevention and Chemotherapy
The induction of O6-alkylGuanine (O6-alkylG) by alkylating agents can result in significant biological consequences such as mutagenicity, cytotoxicity, and carcinogenesis if not repaired by MGMT [13]. The expression of hMGMT in various cultured cell lines has been shown to effectively reduce GC to AT mutations and cytotoxicity caused by alkylating agents [61][62][63][64][65][66]. Several excellent reviews have focused on the role of hMGMT in cancer prevention and chemotherapy [13][24][67][68][69]. N-nitroso compounds, which are present in many foods, play a crucial role in the development of colorectal cancer [69]. The study utilized a transgenic mouse model where mice had high expression of MGMT in the colon [70]. These mice demonstrated reduced rates of aberrant crypt foci (ACF) formation following intraperitoneal administrations of the alkylating agent azoxymethane (AOM). Additionally, the overexpression of MGMT provided protection against G:C to A:T mutations in the KRAS oncogene induced by AOM [70]. Consistent with these findings, depleting MGMT in rats using the potent MGMT inhibitor B6G resulted in an increased frequency of colonic tumors following AOM treatment [71]. MGMT knockout mice, in an inflammation-driven colon carcinogenesis model induced by AOM and dextran sodium sulfate, exhibit high susceptibility to colon tumorigenesis [72][73].
DNA alkylating agents, such as methylating and chloroethylating agents, have been used in cancer therapy for over four decades [67][74][75]. Among these agents, the m6G and O6-chloroethylG products are the main toxic lesions in most cases, particularly at lower doses [75]. However, at higher doses of these agents, N-alkyl purines also contribute to cellular cytotoxicity, and the significance of MGMT in terms of overall cytotoxicity diminishes [75]. The MGMT gene plays a critical role in repairing DNA damage caused by alkylating agents, thereby promoting resistance to chemotherapy. The methylation of the MGMT promoter leads to the reduced or absent expression of hMGMT by inhibiting transcription, thereby increasing sensitivity to alkylating agents [76]. The methylated MGMT promoter has garnered substantial support as a predictive marker for the effectiveness of temozolomide (TMZ), an alkylating agent used in treating glioblastoma and low-grade gliomas. Multiple studies have provided evidence supporting this notion [77][78][79][80][81][82][83][84].
6. The Development of Strategies That Target MGMT
6.1. Non Cancer-Selective MGMT Inhibitors
O6-BG and O6-(4-bromothenyl)guanine (O6-4-BTG) are analogs of m6G that serve as irreversible pseudosubstrates of MGMT [85][86][87][88]. These compounds have been utilized as inhibitors of MGMT in clinical trials [13][75]. O6-BG has gained significant use in sensitizing glioma cells to the alkylating agent TMZ [13][75][89][90]. It possesses the ability to penetrate the blood–brain barrier and deactivate MGMT [24]. Clinical trials combining O6-BG with TMZ have shown promise in delaying brain tumor recurrence and increasing survival time [91][92][93][94]. However, it is important to note that this approach carries an elevated risk of side effects such as hydrocephalus, cerebrospinal fluid leak, and brain infection [95].
Due to the limited water solubility of O6-BG, it is necessary to develop more soluble derivatives to enhance its bioavailability. One approach is the meta-substitution of the aminomethyl group on the benzyl moiety of O6-BG. This modification enhances the water solubility of the compound and results in a more potent inhibitory activity against MGMT [96]. Several other 6-(benzyloxy)pyrimidine derivatives have been synthesized as potential inhibitors of MGMT [13][97]. In addition to the previously mentioned MGMT inhibitors, other types of inhibitors have been discovered. Acrolein and chloromethyltriazoles are highly reactive molecules with nucleophilic sites that can react with cysteine residues, effectively inhibiting MGMT [98]. Another inhibitor, 6-carboxyfluorescein, acts on MGMT in a non-covalent manner [99].
The primary concern regarding the use of non-cancer selective MGMT inhibitors is the increased risk of myelosuppression in bone marrow cells and other normal cells, which can lead to severe hematological toxicity such as leukemia and myelodysplastic syndrome [13][24][97][100]. To address these concerns and for various other reasons, researchers are actively developing cancer-selective inhibitors.
6.2. Cancer-Selective MGMT Inhibitors
The primary strategy for developing a cancer-specific MGMT inhibitor involves modifying the inhibitor with tumor-targeting groups [13][75]. The objective of this approach is to prevent MGMT inhibition in normal tissues while sensitizing cancer cells to chemotherapy [13][75]. Aerobic glycolysis is a prevalent metabolic characteristic observed in numerous tumors. In light of this, the conjugation of a glucose group to the MGMT inhibitor represents a promising concept for the development of cancer-selective MGMT inhibitors [101]. Studies have reported the high efficacy of both O6-BG-Glucose (O6-BG-Glu) and O6-BTG-Glu in inhibiting MGMT in various cancer cell lines, including T98G glioblastoma [88][101][102].
Prodrugs have the potential to enhance tumor specificity and improve pharmacokinetic profiles [13][67]. Many solid tumors exhibit a characteristic of hypoxia, and hypoxia-activated O6-BG prodrugs have already been developed and utilized [103][104]. In particular, the release of β-glucuronidase is commonly observed from necrotic tumor cells. Therefore, designing O6-BG prodrugs that are substrate-related to β-glucuronidase could be a promising strategy to exploit [105].
6.3. Local Drug Delivery
Drug delivery plays a critical role in improving targeted therapy by utilizing diverse delivery systems and strategies to enhance the effectiveness and specificity of therapeutic agents [106]. Local drug delivery can achieve targeted therapy. Gliadel (BCNU wafers) marked the initial clinical application of polymer drug delivery in the treatment of brain tumors. It involves the insertion of BCNU wafers into the resection cavity of patients following surgery [107][108]. These wafers gradually degrade, enabling localized delivery of BCNU to the target area [109]. TMZ was encapsulated within a biologically inert matrix for localized administration to patients with GBM. This encapsulation strategy demonstrated superior efficacy compared to standard therapy alone, resulting in a remarkable increase in overall survival of up to 33 weeks [110]. An injectable enzyme-responsive hydrogel was developed, capable of delivering TMZ and O6-BG. This hydrogel demonstrated effectiveness in reducing the recurrence of TMZ-resistant glioma after surgery, while also enhancing the inhibitory efficiency against tumors [111].
Nanoparticle-based delivery offers unique properties that enable targeted delivery to specific cells or tissues [112]. In a recent study, a combination of O6-BG formulation with a redox-responsive theranostic superparamagnetic iron oxide nanoparticle (SPION) platform was employed to enhance the intracellular delivery of O6-BG to glioblastoma multiforme (GBM) cells while minimizing drug accumulation in healthy tissues [113]. This improved formulation of O6-BG showed a significant decrease in MGMT activity and enhanced the cytotoxic effect of TMZ in vitro. Additionally, in an orthotopic primary human GBM xenograft mouse model, it resulted in a three-fold increase in survival compared to untreated controls [113]. Another formulation involved nanoparticles (NPs) coated with a pH-sensitive polymer and a modified analog of MGMT inhibitor, specifically dialdehyde-modified O6-benzylguanosine (DABGS). This formulation demonstrated a remarkable inhibition of MGMT activity and enhanced the cytotoxicity of TMZ in vitro [114].
6.4. Targeting the Expression of hMGMT
Several strategies have been developed to target the expression of MGMT. Among them, the identification of microRNAs (miRNAs) that regulate MGMT expression by degrading MGMT mRNA shows promise as an innovative treatment approach to enhance TMZ sensitivity in patients with unmethylated MGMT [115]. Notably, miRNAs such as miR-142-3p, miR-181d, miR-370-3p, and others have been discovered to downregulate MGMT expression and enhance sensitivity to TMZ in GBM cell lines [115][116][117][118][119][120][121][122]. Another promising strategy is the use of siRNA to target MGMT. The combination of TMZ with the MGMT–siRNA/liposome complex has shown a strong synergistic antitumor effect [24][123]. A small-molecule compound, EPIC-0412, was discovered to enhance the chemotherapeutic effect of TMZ by epigenetically silencing the expression of MGMT. It achieved this by targeting two key pathways: the p21-E2F1 DNA damage repair axis and the ATF3-p-p65-MGMT axis [124].
6.5. Others
Autoantibodies against MGMT were detected in patients’ serum [125]. The researchers screened the most responsive peptides using these autoantibodies and discovered that these peptides conferred resistance to TMZ in glioma cells both in vivo and in vitro [126]. This finding suggests that monoclonal antibodies targeting these peptides could serve as a novel strategy to overcome resistance in GBM cases with unmethylated MGMT promoters to alkylating agents. The new agent-KL-50 has been found to induce cell killing selectively in MGMT-silenced tumors, independent of mismatch repair (MMR). It creates a dynamic DNA lesion that can be repaired by MGMT. However, in MGMT-deficient conditions, this lesion slowly evolves into an interstrand cross-link, leading to MMR-independent cell death. Notably, this process exhibits low toxicity in both in vitro and in vivo settings [127]. This agent represents a novel approach in designing chemotherapeutics that exploit specific DNA repair defects.
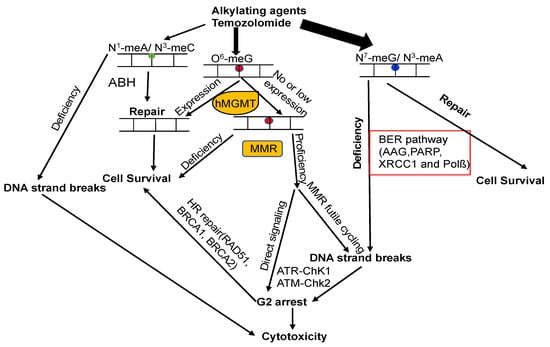
7. The Crosstalk of MGMT with Other DNA Repair Pathways
The attack of alkylating agents on DNA can lead to various types of lesions on the heterocyclic bases or backbone. Repairing methylated DNA adducts involves several pathways, primarily the base excision repair (BER) pathway, the family of AlkB homolog proteins (ALKBH), and MGMT (Figure 5) [5][14][128]. The BER pathway plays a critical role in repairing the main N-alkylation DNA adducts, such as N3-methyladenine, N3-methylguanine, and N7-methylguanine, with the first step of this process involving the alkyladenine-DNA glycosylase (AAG, MPG) [5][14][128][129].
Figure 5. Model of DNA repair pathways involved in DNA alkylation damage. DNA lesions caused by alkylating agents are processed by BER, ALKBH, and MGMT. If m6G is unaffected by MGMT during DNA replication, m6G forms mismatches with T, generating m6G–T pairs. MMR rectifies these mismatches by removing T from m6G–T pairs. This futile cycling replication process results in unreplicated gaps opposite the m6G lesions, which remain tolerated until the subsequent S-phase (middle panel). In this phase, they impede DNA replication, causing DSBs that trigger cell cycle arrest or eventual cell death. Alternatively, mismatch repair proteins recognizing m6G/T lesions might function as a scaffold, directly recruiting DNA damage signaling molecules (middle panel), leading to the activation of cell cycle checkpoints and/or apoptosis. The circles with different color mean different types of DNA lesions.
7.1. MGMT and BER
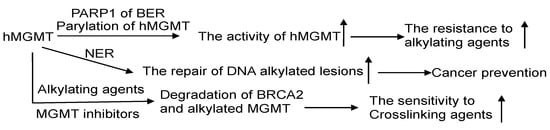
MGMT and BER are the primary pathways to process DNA lesions induced by alkylating agents. As of now, no glycosylase capable of recognizing m6G has been discovered. AAG, on the other hand, specifically recognizes N-alkylated purines and does not compete with MGMT. Nevertheless, there are reports suggesting that MGMT activity is regulated by the BER protein-PARP, which plays a critical role in BER for processing N-alkylpurines [128]. The PARP-mediated PARylation of MGMT, induced by alkylating agents, enhances its binding to chromatin and its ability to facilitate the removal of m6G adducts from DNA (Figure 6) [130][131].
Figure 6. The crosstalk of MGMT with other DNA repair pathways. The interplay of MGMT with other DNA repair mechanisms involves several notable connections. PARP1, a protein in the BER pathway, can perform poly-ADP-ribosylation on hMGMT, resulting in the formation of poly-ADP-ribosylated hMGMT. This modification facilitates the localization of hMGMT within chromatin, enabling efficient repair of DNA lesions. The effectiveness of the NER process can assist MGMT in managing DNA adducts. Through a collaborative effort, MGMT and NER collectively process O6-carboxymethylguanine (O6-CMG) lesions, thereby thwarting potential carcinogenic outcomes. Intriguingly, the alkylation of hMGMT and the existence of alkylating damage can accelerate the degradation of BRCA2, potentially heightening the sensitivity of cancer cells to the treatment of crosslinking agents. ↑: increase.
7.2. MGMT and Nucleotide Excision Repair (NER)
The NER pathway plays a crucial role in repairing bulky helix-distorting DNA adducts, like cyclobutene-pyrimidine dimers induced by UV light [132]. Experimental evidence suggests that cells expressing both MGMT and NER proteins efficiently repair O6-ethylG, indicating a collaborative effort between MGMT and NER in processing this type of DNA damage [133]. The proposed mechanism involves NER proteins opening up the tightly packed chromatin, thereby aiding MGMT in locating and addressing the DNA adducts [133].
7.3. MGMT and MMR
The expression of MGMT plays a critical role in preventing cell death induced by alkylating agents, since it directly converts m6G back to G. Additionally, MMR also has a significant impact on generating cytotoxic effects triggered by m6G lesions [134]. Deficiency in MMR results in resistance against these effects, both in vitro and in vivo [135][136]. Unrepaired m6G is highly stable and tends to pair with Thymine (T) instead of G. This m6G:T mispair is extremely mutagenic, and can be recognized by the MSH2/MSH6 heterodimer of the MMR pathway [137]. The persistence of m6G on the template strand leads to the formation of m6G:T mispairs during MMR-directed strand resynthesis, initiating repeated cycles of futile repair [128][134][138][139]. Consequently, unproductive replication cycles across m6Gs create unreplicated gaps that interfere with DNA replication in the subsequent S-phase, generating double-strand breaks (DSBs) that ultimately cause cell cycle arrest and cell death (Figure 5) [128][134][138][139]. The futile cycle model finds support through in vitro experiments, which demonstrate that the cytotoxicity of m6G takes place during the second cell cycle following treatment [134][138][140]. Circular DNA substrates containing m6G/T mismatches, rather than regular G/T mismatches, elicit a MMR-dependent preferential recruitment of ATR-ATRIP, leading to ATR activation (Figure 5) [141].
7.4. MGMT and DSB Repair
Concomitant with DNA replication, MMR gives rise to DSBs induced by m6G lesions, which are responsible for provoking apoptosis signaling [75][76][128]. Considering the crucial role of DSBs in m6G induced cytotoxicity, DSB repair is expected to significantly impact m6G-induced chromosomal changes. This is supported by the finding that m6G lesions caused higher aberration frequencies in cells with ATM deficiency [142]. Thus, MGMT serves as a key defense against clastogenicity by O6-methylating agents, acting in concert with MMR, BER, and DSB repair (Figure 5) [128]. After DSBs are formed, they undergo repair through homologous recombination (HR) or non-homologous end joining (NHEJ). BRCA2 plays a crucial role in HR and interacts with hMGMT to facilitate the degradation of methylated or benzylated hMGMT [143].
References
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715.
- Friedberg, E.C. Out of the shadows and into the light: The emergence of DNA repair. Trends Biochem. Sci. 1995, 20, 381.
- Pegg, A.E. Repair of O(6)-alkylguanine by alkyltransferases. Mutat. Res. 2000, 462, 83–100.
- Pegg, A.E.; Dolan, M.E.; Moschel, R.C. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase. Prog. Nucleic Acid Res. Mol. Biol. 1995, 51, 167–223.
- Mishina, Y.; Duguid, E.M.; He, C. Direct reversal of DNA alkylation damage. Chem. Rev. 2006, 106, 215–232.
- Hecht, S.S. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat. Res. 1999, 424, 127–142.
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200.
- Rajski, S.R.; Williams, R.M. DNA Cross-Linking Agents as Antitumor Drugs. Chem. Rev. 1998, 98, 2723–2796.
- Sedgwick, B. Repairing DNA-methylation damage. Nat. Rev. Mol. Cell Biol. 2004, 5, 148–157.
- Vaughan, P.; Lindahl, T.; Sedgwick, B. Induction of the adaptive response of Escherichia coli to alkylation damage by the environmental mutagen, methyl chloride. Mutat. Res. 1993, 293, 249–257.
- Barrows, L.R.; Magee, P.N. Nonenzymatic methylation of DNA by S-adenosylmethionine in vitro. Carcinogenesis 1982, 3, 349–351.
- Rydberg, B.; Lindahl, T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J. 1982, 1, 211–216.
- Pegg, A.E. Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem. Res. Toxicol. 2011, 24, 618–639.
- Wyatt, M.D.; Pittman, D.L. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006, 19, 1580–1594.
- Margison, G.P.; Povey, A.C.; Kaina, B.; Santibanez Koref, M.F. Variability and regulation of O6-alkylguanine-DNA alkyltransferase. Carcinogenesis 2003, 24, 625–635.
- Beranek, D.T. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. 1990, 231, 11–30.
- Lindahl, T.; Sedgwick, B.; Sekiguchi, M.; Nakabeppu, Y. Regulation and expression of the adaptive response to alkylating agents. Annu. Rev. Biochem. 1988, 57, 133–157.
- Fornace, A.J., Jr.; Papathanasiou, M.A.; Hollander, M.C.; Yarosh, D.B. Expression of the O6-methylguanine-DNA methyltransferase gene MGMT in MER+ and MER- human tumor cells. Cancer Res. 1990, 50, 7908–7911.
- Bouras, E.; Karakioulaki, M.; Bougioukas, K.I.; Aivaliotis, M.; Tzimagiorgis, G.; Chourdakis, M. Gene promoter methylation and cancer: An umbrella review. Gene 2019, 710, 333–340.
- Kroes, R.A.; Erickson, L.C. The role of mRNA stability and transcription in O6-methylguanine DNA methyltransferase (MGMT) expression in Mer+ human tumor cells. Carcinogenesis 1995, 16, 2255–2257.
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282.
- Costello, J.F.; Futscher, B.W.; Tano, K.; Graunke, D.M.; Pieper, R.O. Graded methylation in the promoter and body of the O6-methylguanine DNA methyltransferase (MGMT) gene correlates with MGMT expression in human glioma cells. J. Biol. Chem. 1994, 269, 17228–17237.
- Watts, G.S.; Pieper, R.O.; Costello, J.F.; Peng, Y.M.; Dalton, W.S.; Futscher, B.W. Methylation of discrete regions of the O6-methylguanine DNA methyltransferase (MGMT) CpG island is associated with heterochromatinization of the MGMT transcription start site and silencing of the gene. Mol. Cell. Biol. 1997, 17, 5612–5619.
- Yu, W.; Zhang, L.; Wei, Q.; Shao, A. O(6)-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Front. Oncol. 2019, 9, 1547.
- Lindahl, T.; Demple, B.; Robins, P. Suicide inactivation of the E. coli O6-methylguanine-DNA methyltransferase. EMBO J. 1982, 1, 1359–1363.
- Olsson, M.; Lindahl, T. Repair of alkylated DNA in Escherichia coli. Methyl group transfer from O6-methylguanine to a protein cysteine residue. J. Biol. Chem. 1980, 255, 10569–10571.
- Samson, L. The suicidal DNA repair methyltransferases of microbes. Mol. Microbiol. 1992, 6, 825–831.
- Samson, L.; Cairns, J. A new pathway for DNA repair in Escherichia coli. Nature 1977, 267, 281–283.
- Potter, P.M.; Wilkinson, M.C.; Fitton, J.; Carr, F.J.; Brennand, J.; Cooper, D.P.; Margison, G.P. Characterisation and nucleotide sequence of ogt, the O6-alkylguanine-DNA-alkyltransferase gene of E. coli. Nucleic Acids Res. 1987, 15, 9177–9193.
- Rebeck, G.W.; Smith, C.M.; Goad, D.L.; Samson, L. Characterization of the major DNA repair methyltransferase activity in unadapted Escherichia coli and identification of a similar activity in Salmonella typhimurium. J. Bacteriol. 1989, 171, 4563–4568.
- Wibley, J.E.; Pegg, A.E.; Moody, P.C. Crystal structure of the human O(6)-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 2000, 28, 393–401.
- Daniels, D.S.; Mol, C.D.; Arvai, A.S.; Kanugula, S.; Pegg, A.E.; Tainer, J.A. Active and alkylated human AGT structures: A novel zinc site, inhibitor and extrahelical base binding. EMBO J. 2000, 19, 1719–1730.
- Daniels, D.S.; Woo, T.T.; Luu, K.X.; Noll, D.M.; Clarke, N.D.; Pegg, A.E.; Tainer, J.A. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat. Struct. Mol. Biol. 2004, 11, 714–720.
- Duguid, E.M.; Rice, P.A.; He, C. The structure of the human AGT protein bound to DNA and its implications for damage detection. J. Mol. Biol. 2005, 350, 657–666.
- Fang, Q.; Kanugula, S.; Pegg, A.E. Function of domains of human O6-alkylguanine-DNA alkyltransferase. Biochemistry 2005, 44, 15396–15405.
- Guengerich, F.P.; Fang, Q.; Liu, L.; Hachey, D.L.; Pegg, A.E. O6-alkylguanine-DNA alkyltransferase: Low pKa and high reactivity of cysteine 145. Biochemistry 2003, 42, 10965–10970.
- Demple, B.; Sedgwick, B.; Robins, P.; Totty, N.; Waterfield, M.D.; Lindahl, T. Active site and complete sequence of the suicidal methyltransferase that counters alkylation mutagenesis. Proc. Natl. Acad. Sci. USA 1985, 82, 2688–2692.
- Sedgwick, B.; Robins, P.; Totty, N.; Lindahl, T. Functional domains and methyl acceptor sites of the Escherichia coli ada protein. J. Biol. Chem. 1988, 263, 4430–4433.
- He, C.; Hus, J.C.; Sun, L.J.; Zhou, P.; Norman, D.P.; Dotsch, V.; Wei, H.; Gross, J.D.; Lane, W.S.; Wagner, G.; et al. A methylation-dependent electrostatic switch controls DNA repair and transcriptional activation by E. coli ada. Mol. Cell 2005, 20, 117–129.
- Teo, I.; Sedgwick, B.; Kilpatrick, M.W.; McCarthy, T.V.; Lindahl, T. The intracellular signal for induction of resistance to alkylating agents in E. coli. Cell 1986, 45, 315–324.
- Akimaru, H.; Sakumi, K.; Yoshikai, T.; Anai, M.; Sekiguchi, M. Positive and negative regulation of transcription by a cleavage product of Ada protein. J. Mol. Biol. 1990, 216, 261–273.
- Landini, P.; Busby, S.J. The Escherichia coli Ada protein can interact with two distinct determinants in the sigma70 subunit of RNA polymerase according to promoter architecture: Identification of the target of Ada activation at the alkA promoter. J. Bacteriol. 1999, 181, 1524–1529.
- Landini, P.; Volkert, M.R. Transcriptional activation of the Escherichia coli adaptive response gene aidB is mediated by binding of methylated Ada protein. Evidence for a new consensus sequence for Ada-binding sites. J. Biol. Chem. 1995, 270, 8285–8289.
- Myers, L.C.; Terranova, M.P.; Ferentz, A.E.; Wagner, G.; Verdine, G.L. Repair of DNA methylphosphotriesters through a metalloactivated cysteine nucleophile. Science 1993, 261, 1164–1167.
- Myers, L.C.; Jackow, F.; Verdine, G.L. Metal dependence of transcriptional switching in Escherichia coli Ada. J. Biol. Chem. 1995, 270, 6664–6670.
- Myers, L.C.; Terranova, M.P.; Nash, H.M.; Markus, M.A.; Verdine, G.L. Zinc binding by the methylation signaling domain of the Escherichia coli Ada protein. Biochemistry 1992, 31, 4541–4547.
- Daniels, D.S.; Tainer, J.A. Conserved structural motifs governing the stoichiometric repair of alkylated DNA by O(6)-alkylguanine-DNA alkyltransferase. Mutat. Res. 2000, 460, 151–163.
- Moore, M.H.; Gulbis, J.M.; Dodson, E.J.; Demple, B.; Moody, P.C. Crystal structure of a suicidal DNA repair protein: The Ada O6-methylguanine-DNA methyltransferase from E. coli. EMBO J. 1994, 13, 1495–1501.
- Paalman, S.R.; Sung, C.; Clarke, N.D. Specificity of DNA repair methyltransferases determined by competitive inactivation with oligonucleotide substrates: Evidence that Escherichia coli Ada repairs O6-methylguanine and O4-methylthymine with similar efficiency. Biochemistry 1997, 36, 11118–11124.
- Pegg, A.E.; Wiest, L.; Mummert, C.; Stine, L.; Moschel, R.C.; Dolan, M.E. Use of antibodies to human O6-alkylguanine-DNA alkyltransferase to study the content of this protein in cells treated with O6-benzylguanine or N-methyl-N′-nitro-N-nitrosoguanidine. Carcinogenesis 1991, 12, 1679–1683.
- Srivenugopal, K.S.; Yuan, X.H.; Friedman, H.S.; Ali-Osman, F. Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemistry 1996, 35, 1328–1334.
- Xu-Welliver, M.; Pegg, A.E. Degradation of the alkylated form of the DNA repair protein, O(6)-alkylguanine-DNA alkyltransferase. Carcinogenesis 2002, 23, 823–830.
- Edara, S.; Kanugula, S.; Pegg, A.E. Expression of the inactive C145A mutant human O6-alkylguanine-DNA alkyltransferase in E. coli increases cell killing and mutations by N-methyl-N′-nitro-N-nitrosoguanidine. Carcinogenesis 1999, 20, 103–108.
- Rasimas, J.J.; Dalessio, P.A.; Ropson, I.J.; Pegg, A.E.; Fried, M.G. Active-site alkylation destabilizes human O6-alkylguanine DNA alkyltransferase. Protein Sci. Publ. Protein Soc. 2004, 13, 301–305.
- Rasimas, J.J.; Pegg, A.E.; Fried, M.G. DNA-binding mechanism of O6-alkylguanine-DNA alkyltransferase. Effects of protein and DNA alkylation on complex stability. J. Biol. Chem. 2003, 278, 7973–7980.
- Ali, R.B.; Teo, A.K.; Oh, H.K.; Chuang, L.S.; Ayi, T.C.; Li, B.F. Implication of localization of human DNA repair enzyme O6-methylguanine-DNA methyltransferase at active transcription sites in transcription-repair coupling of the mutagenic O6-methylguanine lesion. Mol. Cell. Biol. 1998, 18, 1660–1669.
- Teo, A.K.; Oh, H.K.; Ali, R.B.; Li, B.F. The modified human DNA repair enzyme O(6)-methylguanine-DNA methyltransferase is a negative regulator of estrogen receptor-mediated transcription upon alkylation DNA damage. Mol. Cell. Biol. 2001, 21, 7105–7114.
- Wu, X.; Luo, Q.; Zhao, P.; Chang, W.; Wang, Y.; Shu, T.; Ding, F.; Li, B.; Liu, Z. MGMT-activated DUB3 stabilizes MCL1 and drives chemoresistance in ovarian cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 2961–2966.
- Horiguchi, M.; Kim, J.; Matsunaga, N.; Kaji, H.; Egawa, T.; Makino, K.; Koyanagi, S.; Ohdo, S. Glucocorticoid-dependent expression of O(6)-methylguanine-DNA methyltransferase gene modulates dacarbazine-induced hepatotoxicity in mice. J. Pharmacol. Exp. Ther. 2010, 333, 782–787.
- Pegg, A.E.; Perry, W.; Bennett, R.A. Effect of partial hepatectomy on removal of O6-methylguanine from alkylated DNA by rat liver extracts. Biochem. J. 1981, 197, 195–201.
- Shrivastav, N.; Li, D.; Essigmann, J.M. Chemical biology of mutagenesis and DNA repair: Cellular responses to DNA alkylation. Carcinogenesis 2010, 31, 59–70.
- Choi, J.Y.; Chowdhury, G.; Zang, H.; Angel, K.C.; Vu, C.C.; Peterson, L.A.; Guengerich, F.P. Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 2006, 281, 38244–38256.
- Loechler, E.L.; Green, C.L.; Essigmann, J.M. In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc. Natl. Acad. Sci. USA 1984, 81, 6271–6275.
- Pence, M.G.; Choi, J.Y.; Egli, M.; Guengerich, F.P. Structural basis for proficient incorporation of dTTP opposite O6-methylguanine by human DNA polymerase iota. J. Biol. Chem. 2010, 285, 40666–40672.
- Warren, J.J.; Forsberg, L.J.; Beese, L.S. The structural basis for the mutagenicity of O(6)-methyl-guanine lesions. Proc. Natl. Acad. Sci. USA 2006, 103, 19701–19706.
- Woodside, A.M.; Guengerich, F.P. Effect of the O6 substituent on misincorporation kinetics catalyzed by DNA polymerases at O(6)-methylguanine and O(6)-benzylguanine. Biochemistry 2002, 41, 1027–1038.
- Bai, P.; Fan, T.; Sun, G.; Wang, X.; Zhao, L.; Zhong, R. The dual role of DNA repair protein MGMT in cancer prevention and treatment. DNA Repair 2023, 123, 103449.
- Butler, M.; Pongor, L.; Su, Y.T.; Xi, L.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391.
- Fahrer, J.; Kaina, B. O6-methylguanine-DNA methyltransferase in the defense against N-nitroso compounds and colorectal cancer. Carcinogenesis 2013, 34, 2435–2442.
- Zaidi, N.H.; Pretlow, T.P.; O’Riordan, M.A.; Dumenco, L.L.; Allay, E.; Gerson, S.L. Transgenic expression of human MGMT protects against azoxymethane-induced aberrant crypt foci and G to A mutations in the K-ras oncogene of mouse colon. Carcinogenesis 1995, 16, 451–456.
- Wali, R.K.; Skarosi, S.; Hart, J.; Zhang, Y.; Dolan, M.E.; Moschel, R.C.; Nguyen, L.; Mustafi, R.; Brasitus, T.A.; Bissonnette, M. Inhibition of O(6)-methylguanine-DNA methyltransferase increases azoxymethane-induced colonic tumors in rats. Carcinogenesis 1999, 20, 2355–2360.
- Wirtz, S.; Nagel, G.; Eshkind, L.; Neurath, M.F.; Samson, L.D.; Kaina, B. Both base excision repair and O6-methylguanine-DNA methyltransferase protect against methylation-induced colon carcinogenesis. Carcinogenesis 2010, 31, 2111–2117.
- Bugni, J.M.; Meira, L.B.; Samson, L.D. Alkylation-induced colon tumorigenesis in mice deficient in the Mgmt and Msh6 proteins. Oncogene 2009, 28, 734–741.
- Gerson, S.L. MGMT: Its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer 2004, 4, 296–307.
- Kaina, B.; Margison, G.P.; Christmann, M. Targeting O(6)-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell. Mol. Life Sci. 2010, 67, 3663–3681.
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair 2019, 78, 128–141.
- Hegi, M.E.; Diserens, A.C.; Godard, S.; Dietrich, P.Y.; Regli, L.; Ostermann, S.; Otten, P.; Van Melle, G.; de Tribolet, N.; Stupp, R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874.
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003.
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354.
- Aoki, K.; Natsume, A. Overview of DNA methylation in adult diffuse gliomas. Brain Tumor Pathol. 2019, 36, 84–91.
- Reifenberger, G.; Hentschel, B.; Felsberg, J.; Schackert, G.; Simon, M.; Schnell, O.; Westphal, M.; Wick, W.; Pietsch, T.; Loeffler, M.; et al. Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. Int. J. Cancer 2012, 131, 1342–1350.
- Qi, F.; Yin, Z.; Wang, G.; Zeng, S. Clinical and Prognostic Significance of O(6)-Methylguanine-DNA Methyltransferase Promoter Methylation in Patients with Melanoma: A Systematic Meta-Analysis. Ann. Dermatol. 2018, 30, 129–135.
- Pandith, A.A.; Qasim, I.; Zahoor, W.; Shah, P.; Bhat, A.R.; Sanadhya, D.; Shah, Z.A.; Naikoo, N.A. Concordant association validates MGMT methylation and protein expression as favorable prognostic factors in glioma patients on alkylating chemotherapy (Temozolomide). Sci. Rep. 2018, 8, 6704.
- Dahlrot, R.H.; Larsen, P.; Boldt, H.B.; Kreutzfeldt, M.S.; Hansen, S.; Hjelmborg, J.B.; Kristensen, B.W. Posttreatment Effect of MGMT Methylation Level on Glioblastoma Survival. J. Neuropathol. Exp. Neurol. 2019, 78, 633–640.
- Dolan, M.E.; Pegg, A.E. O6-benzylguanine and its role in chemotherapy. Clin. Cancer Res. 1997, 3, 837–847.
- Dolan, M.E.; Moschel, R.C.; Pegg, A.E. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc. Natl. Acad. Sci. USA 1990, 87, 5368–5372.
- Shibata, T.; Glynn, N.; McMurry, T.B.; McElhinney, R.S.; Margison, G.P.; Williams, D.M. Novel synthesis of O6-alkylguanine containing oligodeoxyribonucleotides as substrates for the human DNA repair protein, O6-methylguanine DNA methyltransferase (MGMT). Nucleic Acids Res. 2006, 34, 1884–1891.
- Kaina, B.; Muhlhausen, U.; Piee-Staffa, A.; Christmann, M.; Garcia Boy, R.; Rosch, F.; Schirrmacher, R. Inhibition of O6-methylguanine-DNA methyltransferase by glucose-conjugated inhibitors: Comparison with nonconjugated inhibitors and effect on fotemustine and temozolomide-induced cell death. J. Pharmacol. Exp. Ther. 2004, 311, 585–593.
- Middleton, M.R.; Margison, G.P. Improvement of chemotherapy efficacy by inactivation of a DNA-repair pathway. Lancet Oncol. 2003, 4, 37–44.
- Bobola, M.S.; Silber, J.R.; Ellenbogen, R.G.; Geyer, J.R.; Blank, A.; Goff, R.D. O6-methylguanine-DNA methyltransferase, O6-benzylguanine, and resistance to clinical alkylators in pediatric primary brain tumor cell lines. Clin. Cancer Res. 2005, 11, 2747–2755.
- Koch, D.; Hundsberger, T.; Boor, S.; Kaina, B. Local intracerebral administration of O(6)-benzylguanine combined with systemic chemotherapy with temozolomide of a patient suffering from a recurrent glioblastoma. J. Neurooncol. 2007, 82, 85–89.
- Broniscer, A.; Gururangan, S.; MacDonald, T.J.; Goldman, S.; Packer, R.J.; Stewart, C.F.; Wallace, D.; Danks, M.K.; Friedman, H.S.; Poussaint, T.Y.; et al. Phase I trial of single-dose temozolomide and continuous administration of O6-benzylguanine in children with brain tumors: A pediatric brain tumor consortium report. Clin. Cancer Res. 2007, 13, 6712–6718.
- Quinn, J.A.; Desjardins, A.; Weingart, J.; Brem, H.; Dolan, M.E.; Delaney, S.M.; Vredenburgh, J.; Rich, J.; Friedman, A.H.; Reardon, D.A.; et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J. Clin. Oncol. 2005, 23, 7178–7187.
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase I trial of temozolomide plus O6-benzylguanine 5-day regimen with recurrent malignant glioma. Neuro-Oncology 2009, 11, 556–561.
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase II trial of temozolomide plus O6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J. Clin. Oncol. 2009, 27, 1262–1267.
- Pauly, G.T.; Loktionova, N.A.; Fang, Q.; Vankayala, S.L.; Guida, W.C.; Pegg, A.E. Substitution of aminomethyl at the meta-position enhances the inactivation of O6-alkylguanine-DNA alkyltransferase by O6-benzylguanine. J. Med. Chem. 2008, 51, 7144–7153.
- Sun, G.; Zhao, L.; Zhong, R.; Peng, Y. The specific role of O(6)-methylguanine-DNA methyltransferase inhibitors in cancer chemotherapy. Future Med. Chem. 2018, 10, 1971–1996.
- Wang, C.; Abegg, D.; Hoch, D.G.; Adibekian, A. Chemoproteomics-Enabled Discovery of a Potent and Selective Inhibitor of the DNA Repair Protein MGMT. Angew. Chem. Int. Ed. 2016, 55, 2911–2915.
- Melikishvili, M.; Rodgers, D.W.; Fried, M.G. 6-Carboxyfluorescein and structurally similar molecules inhibit DNA binding and repair by O(6)-alkylguanine DNA alkyltransferase. DNA Repair 2011, 10, 1193–1202.
- Geiger, H.; Schleimer, D.; Nattamai, K.J.; Dannenmann, S.R.; Davies, S.M.; Weiss, B.D. Mutagenic potential of temozolomide in bone marrow cells in vivo. Blood 2006, 107, 3010–3011.
- Reinhard, J.; Eichhorn, U.; Wiessler, M.; Kaina, B. Inactivation of O(6)-methylguanine-DNA methyltransferase by glucose-conjugated inhibitors. Int. J. Cancer 2001, 93, 373–379.
- Tomaszowski, K.H.; Schirrmacher, R.; Kaina, B. Multidrug Efflux Pumps Attenuate the Effect of MGMT Inhibitors. Mol. Pharm. 2015, 12, 3924–3934.
- Zhu, R.; Baumann, R.P.; Penketh, P.G.; Shyam, K.; Sartorelli, A.C. Hypoxia-selective O6-alkylguanine-DNA alkyltransferase inhibitors: Design, synthesis, and evaluation of 6-(benzyloxy)-2-(aryldiazenyl)-9H-purines as prodrugs of O6-benzylguanine. J. Med. Chem. 2013, 56, 1355–1359.
- Zhu, R.; Liu, M.C.; Luo, M.Z.; Penketh, P.G.; Baumann, R.P.; Shyam, K.; Sartorelli, A.C. 4-nitrobenzyloxycarbonyl derivatives of O(6)-benzylguanine as hypoxia-activated prodrug inhibitors of O(6)-alkylguanine-DNA alkyltransferase (AGT), which produces resistance to agents targeting the O-6 position of DNA guanine. J. Med. Chem. 2011, 54, 7720–7728.
- Wei, G.; Loktionova, N.A.; Pegg, A.E.; Moschel, R.C. Beta-glucuronidase-cleavable prodrugs of O6-benzylguanine and O6-benzyl-2′-deoxyguanosine. J. Med. Chem. 2005, 48, 256–261.
- Tseng, Y.Y.; Kau, Y.C.; Liu, S.J. Advanced interstitial chemotherapy for treating malignant glioma. Expert Opin. Drug Deliv. 2016, 13, 1533–1544.
- Guerin, C.; Olivi, A.; Weingart, J.D.; Lawson, H.C.; Brem, H. Recent advances in brain tumor therapy: Local intracerebral drug delivery by polymers. Investig. New Drugs 2004, 22, 27–37.
- Weinberg, B.D.; Blanco, E.; Gao, J. Polymer implants for intratumoral drug delivery and cancer therapy. J. Pharm. Sci. 2008, 97, 1681–1702.
- Weingart, J.; Grossman, S.A.; Carson, K.A.; Fisher, J.D.; Delaney, S.M.; Rosenblum, M.L.; Olivi, A.; Judy, K.; Tatter, S.B.; Dolan, M.E. Phase I trial of polifeprosan 20 with carmustine implant plus continuous infusion of intravenous O6-benzylguanine in adults with recurrent malignant glioma: New approaches to brain tumor therapy CNS consortium trial. J. Clin. Oncol. 2007, 25, 399–404.
- Karlsson, I.; Veevnik, D.; Fedulov, A.; Yurkshtovich, N.; Yurkshtovich, T.; Pejler, G.; Lokot, I. Local delivery of temozolomide via a biologically inert carrier (Temodex) prolongs survival in glioma patients, irrespectively of the methylation status of MGMT. Neoplasma 2019, 66, 288–293.
- Zhao, Z.; Shen, J.; Zhang, L.; Wang, L.; Xu, H.; Han, Y.; Jia, J.; Lu, Y.; Yu, R.; Liu, H. Injectable postoperative enzyme-responsive hydrogels for reversing temozolomide resistance and reducing local recurrence after glioma operation. Biomater. Sci. 2020, 8, 5306–5316.
- Kievit, F.M.; Wang, F.Y.; Fang, C.; Mok, H.; Wang, K.; Silber, J.R.; Ellenbogen, R.G.; Zhang, M. Doxorubicin loaded iron oxide nanoparticles overcome multidrug resistance in cancer in vitro. J. Control. Release 2011, 152, 76–83.
- Stephen, Z.R.; Kievit, F.M.; Veiseh, O.; Chiarelli, P.A.; Fang, C.; Wang, K.; Hatzinger, S.J.; Ellenbogen, R.G.; Silber, J.R.; Zhang, M. Redox-responsive magnetic nanoparticle for targeted convection-enhanced delivery of O6-benzylguanine to brain tumors. ACS Nano 2014, 8, 10383–10395.
- Stephen, Z.R.; Gebhart, R.N.; Jeon, M.; Blair, A.A.; Ellenbogen, R.G.; Silber, J.R.; Zhang, M. pH-Sensitive O6-Benzylguanosine Polymer Modified Magnetic Nanoparticles for Treatment of Glioblastomas. Bioconjug. Chem. 2017, 28, 194–202.
- Kirstein, A.; Schmid, T.E.; Combs, S.E. The Role of miRNA for the Treatment of MGMT Unmethylated Glioblastoma Multiforme. Cancers 2020, 12, 1099.
- Cabrini, G.; Fabbri, E.; Lo Nigro, C.; Dechecchi, M.C.; Gambari, R. Regulation of expression of O6-methylguanine-DNA methyltransferase and the treatment of glioblastoma (Review). Int. J. Oncol. 2015, 47, 417–428.
- Chiou, G.Y.; Chien, C.S.; Wang, M.L.; Chen, M.T.; Yang, Y.P.; Yu, Y.L.; Chien, Y.; Chang, Y.C.; Shen, C.C.; Chio, C.C.; et al. Epigenetic regulation of the miR142-3p/interleukin-6 circuit in glioblastoma. Mol. Cell 2013, 52, 693–706.
- Lee, Y.Y.; Yarmishyn, A.A.; Wang, M.L.; Chen, H.Y.; Chiou, S.H.; Yang, Y.P.; Lin, C.F.; Huang, P.I.; Chen, Y.W.; Ma, H.I.; et al. MicroRNA-142-3p is involved in regulation of MGMT expression in glioblastoma cells. Cancer Manag. Res. 2018, 10, 775–785.
- Gao, Y.T.; Chen, X.B.; Liu, H.L. Up-regulation of miR-370-3p restores glioblastoma multiforme sensitivity to temozolomide by influencing MGMT expression. Sci. Rep. 2016, 6, 32972.
- Nadaradjane, A.; Briand, J.; Bougras-Cartron, G.; Disdero, V.; Vallette, F.M.; Frenel, J.S.; Cartron, P.F. miR-370-3p Is a Therapeutic Tool in Anti-glioblastoma Therapy but Is Not an Intratumoral or Cell-free Circulating Biomarker. Mol. Ther. Nucleic Acids 2018, 13, 642–650.
- Quintavalle, C.; Mangani, D.; Roscigno, G.; Romano, G.; Diaz-Lagares, A.; Iaboni, M.; Donnarumma, E.; Fiore, D.; De Marinis, P.; Soini, Y.; et al. MiR-221/222 target the DNA methyltransferase MGMT in glioma cells. PLoS ONE 2013, 8, e74466.
- Zhang, W.; Zhang, J.; Hoadley, K.; Kushwaha, D.; Ramakrishnan, V.; Li, S.; Kang, C.; You, Y.; Jiang, C.; Song, S.W.; et al. miR-181d: A predictive glioblastoma biomarker that downregulates MGMT expression. Neuro-Oncology 2012, 14, 712–719.
- Kato, T.; Natsume, A.; Toda, H.; Iwamizu, H.; Sugita, T.; Hachisu, R.; Watanabe, R.; Yuki, K.; Motomura, K.; Bankiewicz, K.; et al. Efficient delivery of liposome-mediated MGMT-siRNA reinforces the cytotoxity of temozolomide in GBM-initiating cells. Gene Ther. 2010, 17, 1363–1371.
- Zhao, J.; Yang, S.; Cui, X.; Wang, Q.; Yang, E.; Tong, F.; Hong, B.; Xiao, M.; Xin, L.; Xu, C.; et al. A novel compound EPIC-0412 reverses temozolomide resistance via inhibiting DNA repair/MGMT in glioblastoma. Neuro-Oncology 2023, 25, 857–870.
- Wu, H.; Deng, Z.; Wang, H.; Li, X.; Sun, T.; Tao, Z.; Yao, L.; Jin, Y.; Wang, X.; Yang, L.; et al. MGMT autoantibodies as a potential prediction of recurrence and treatment response biomarker for glioma patients. Cancer Med. 2019, 8, 4359–4369.
- Wu, Y.; Zhang, K.; Wang, H.; Chen, G.; Liu, Y.; Li, W.; Zhou, Y. Experimental study of selective MGMT peptides mimicking TMZ drug resistance in glioma. Biochem. Biophys. Rep. 2022, 32, 101386.
- Lin, K.; Gueble, S.E.; Sundaram, R.K.; Huseman, E.D.; Bindra, R.S.; Herzon, S.B. Mechanism-based design of agents that selectively target drug-resistant glioma. Science 2022, 377, 502–511.
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099.
- Serrano-Heras, G.; Castro-Robles, B.; Romero-Sanchez, C.M.; Carrion, B.; Barbella-Aponte, R.; Sandoval, H.; Segura, T. Involvement of N-methylpurine DNA glycosylase in resistance to temozolomide in patient-derived glioma cells. Sci. Rep. 2020, 10, 22185.
- Cropper, J.D.; Alimbetov, D.S.; Brown, K.T.G.; Likhotvorik, R.I.; Robles, A.J.; Guerra, J.T.; He, B.; Chen, Y.; Kwon, Y.; Kurmasheva, R.T. PARP1-MGMT complex underpins pathway crosstalk in O(6)-methylguanine repair. J. Hematol. Oncol. 2022, 15, 146.
- Wu, S.; Li, X.; Gao, F.; de Groot, J.F.; Koul, D.; Yung, W.K.A. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro Oncol. 2021, 23, 920–931.
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481.
- Bronstein, S.M.; Skopek, T.R.; Swenberg, J.A. Efficient repair of O6-ethylguanine, but not O4-ethylthymine or O2-ethylthymine, is dependent upon O6-alkylguanine-DNA alkyltransferase and nucleotide excision repair activities in human cells. Cancer Res. 1992, 52, 2008–2011.
- Gupta, D.; Heinen, C.D. The mismatch repair-dependent DNA damage response: Mechanisms and implications. DNA Repair 2019, 78, 60–69.
- Fink, D.; Aebi, S.; Howell, S.B. The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 1998, 4, 1–6.
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629.
- Duckett, D.R.; Drummond, J.T.; Murchie, A.I.; Reardon, J.T.; Sancar, A.; Lilley, D.M.; Modrich, P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc. Natl. Acad. Sci. USA 1996, 93, 6443–6447.
- Mojas, N.; Lopes, M.; Jiricny, J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 2007, 21, 3342–3355.
- Kaina, B.; Ziouta, A.; Ochs, K.; Coquerelle, T. Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex-, Mex+ and methylation-tolerant mismatch repair compromised cells: Facts and models. Mutat. Res. 1997, 381, 227–241.
- Karran, P.; Marinus, M.G. Mismatch correction at O6-methylguanine residues in E. coli DNA. Nature 1982, 296, 868–869.
- Yoshioka, K.; Yoshioka, Y.; Hsieh, P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol. Cell 2006, 22, 501–510.
- Debiak, M.; Nikolova, T.; Kaina, B. Loss of ATM sensitizes against O6-methylguanine triggered apoptosis, SCEs and chromosomal aberrations. DNA Repair 2004, 3, 359–368.
- Philip, S.; Swaminathan, S.; Kuznetsov, S.G.; Kanugula, S.; Biswas, K.; Chang, S.; Loktionova, N.A.; Haines, D.C.; Kaldis, P.; Pegg, A.E.; et al. Degradation of BRCA2 in alkyltransferase-mediated DNA repair and its clinical implications. Cancer Res. 2008, 68, 9973–9981.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
569
Revisions:
2 times
(View History)
Update Date:
22 Jan 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No