Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elisabet Uribe-Carretero | -- | 3709 | 2024-01-09 12:51:50 | | | |
| 2 | Sirius Huang | + 11 word(s) | 3720 | 2024-01-10 02:21:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Uribe-Carretero, E.; Rey, V.; Fuentes, J.M.; Tamargo-Gómez, I. Lysosomal Storage Disorders Linked to Impaired Autophagy. Encyclopedia. Available online: https://encyclopedia.pub/entry/53608 (accessed on 26 July 2026).
Uribe-Carretero E, Rey V, Fuentes JM, Tamargo-Gómez I. Lysosomal Storage Disorders Linked to Impaired Autophagy. Encyclopedia. Available at: https://encyclopedia.pub/entry/53608. Accessed July 26, 2026.
Uribe-Carretero, Elisabet, Verónica Rey, Jose Manuel Fuentes, Isaac Tamargo-Gómez. "Lysosomal Storage Disorders Linked to Impaired Autophagy" Encyclopedia, https://encyclopedia.pub/entry/53608 (accessed July 26, 2026).
Uribe-Carretero, E., Rey, V., Fuentes, J.M., & Tamargo-Gómez, I. (2024, January 09). Lysosomal Storage Disorders Linked to Impaired Autophagy. In Encyclopedia. https://encyclopedia.pub/entry/53608
Uribe-Carretero, Elisabet, et al. "Lysosomal Storage Disorders Linked to Impaired Autophagy." Encyclopedia. Web. 09 January, 2024.
Copy Citation
Lysosomes are the main organelles responsible for the degradation of macromolecules in eukaryotic cells. Beyond their fundamental role in degradation, lysosomes are involved in different physiological processes such as autophagy, nutrient sensing, and intracellular signaling. In some circumstances, lysosomal abnormalities underlie several human pathologies with different etiologies known as Lysosomal Storage Disorders (LSDs). These disorders can result from deficiencies in primary lysosomal enzymes, dysfunction of lysosomal enzyme activators, alterations in modifiers that impact lysosomal function, or changes in membrane-associated proteins, among other factors.
autophagy
lysosome
autophagosome
lysosomal storage disease
genetic mutations
1. Introduction
Lysosomal storage disorders (LSDs) arise from abnormal lysosomal function, leading to the accumulation of undegraded metabolites. The specific composition of these materials accumulated in lysosomes varies significantly among LSDs. Given the relevance of the lysosomal pathway in cellular homeostasis, its malfunction leads to the dysregulation of several cellular processes linked to this organelle such as lipid homeostasis, cell viability, exocytosis, membrane repair, and autophagy, among others [1]. Lysosomes are the final scenario of autophagic degradation, and the inability to clear autophagosomes results in the accumulation of undesired cargo that can further hinder cell viability. Thus, it is not surprising that defective autophagy has already been described in different LSDs [2], contributing to the development of the disease.
2. Mucopolysaccharidoses
Mucopolysaccharidoses (MPS) are a group of inherited diseases caused by mutations in genes encoding several lysosomal enzymes (including ARSB, GALNS, GLB1, GNS, GUSB, HGSNAT, HYAL1, IDS, IDUA, NAGLU, and SGSH), which are involved in the degradation of glycosaminoglycans (GAGs). However, it was described that additional alterations in other pathways (including autophagy) play a significant role in the pathogenesis of these diseases [3]. In this regard, GAGs are linear polysaccharides composed of repeating disaccharide units, which are highly sulphated. This heterogeneous group includes molecules like hyaluronic acid, heparan sulphate, dermatan sulphate, or keratan sulphate, and they are present in all human tissues. Therefore, accumulation of GAGs affects a wide range of organs and systems, and the CNS is the most commonly impacted. MPS patients exhibit a range of symptoms that differ in severity but share certain characteristic traits. These features involve facial characteristics, skeletal abnormalities, and multiorganic affectation such as heart issues, respiratory problems, and the enlargement of the liver and spleen. Thirteen different types and subtypes of MPSs have been described: MPS I (which includes three subtypes: Hurler, Hurler–Scheie, and Scheie syndromes), MPS II (also known as Hunter syndrome), MPS III (subtypes IIIA, IIIB, IIIC, and IIID), MPS IV (subtypes IVA and IVB), MPS VI, MPS VII, and MPS IX [4]. Mutations related to NCLs are listed in Supplementary Table S1 (can be downloaded at: https://www.mdpi.com/article/10.3390/biology13010034/s1) [5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92].
From a mechanistic point of view, autophagy levels were recently addressed in different models of MPS, resulting in conflicting conclusions [93]. For example, impaired autophagy was described or suggested in MPS II [94], MPS III-A [95], MPS III-B [96], MPS IV-A [97], MPS VI [98], and MPS VII [99]. However, other reports also showed unaltered autophagy in MPS I and III-B [100][101] or even enhanced autophagic response in MPS I, II, and VII [102][103]. Though we cannot rule out the possibility of differential autophagy regulation in MPS subtypes, a possible explanation for this controversy could be the lack of complete, comprehensive autophagy flux analyses, which hinders the precise characterization of the autophagic pathway in these diseases, as has already been discussed in MPS III-C [104]. Interestingly, Kondo and collaborators described a new MPS-like disorder caused by a mutation in VPS33A [105], a gene that encodes for a protein that mediates autophagosome–lysosome fusion. However, this mutation (c.1492C > T; p.Arg498Trp) does not compromise the autophagy-related role of VPS33A, unveiling a new function of this protein (Figure 1: mucopolysaccharidoses).
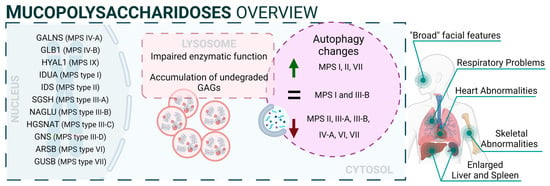
Figure 1. Mucopolysaccharidoses overview.
3. Autophagy in Glycogenoses
The glycogenoses are LSDs characterized by severe autophagy defects specifically affecting skeletal and cardiac muscles. In this regard, these diseases are commonly referred to as autophagic vacuolar myopathies (AVMs), although neuropathological manifestations are also observed in some particular cases [106]. Conditions leading to AVM are associated with genetic mutations affecting genes responsible for glycogen hydrolysis, lysosome acidification, and the maturation and fusion of autophagosomes with lysosomes [107]. Mutations related to glycogenoses are listed in Supplementary Table S2 [108][109][110][111][112][113][114][115][116][117][118][119][120][121][122][123][124][125][126][127][128][129][130][131][132][133][134][135][136][137].
3.1. Pompe Disease
Pompe disease is a rare disorder caused by autosomal recessive mutations in the GAA gene that result in the accumulation of glycogen within lysosomes [138][139]. It was the first recognized LSD [140] and is also known as glycogen storage disease type II (GSDII). Different studies established an association between specific GAA single-nucleotide polymorphisms (SNPs) and Pompe disease, including rs140826989, rs121907938, rs121907945, rs121907936, rs147804176, rs1555600061, rs1800312, and rs200856561, among others [116][131][134]. Clinically, Pompe disease includes two main groups: infantile-onset Pompe disease (IOPD) and late-onset Pompe disease (LOPD) [141]. IOPD patients typically exhibit severe symptoms within the first few months of life, including weakness, developmental delay, feeding difficulties, hypotonia, macroglossia, and hypertrophic cardiomyopathy. These patients experience multisystem glycogen accumulation with less than 1% of normal GAA enzyme function. Without treatment, their life expectancy is typically less than two years [142].
On the other hand, LOPD patients may present symptoms anywhere from childhood to adulthood, with worse prognosis when symptoms manifest at an earlier age. These patients generally retain 1–20% of normal GAA function. However, abnormal glycogen build-up in the respiratory system can lead to respiratory failure, necessitating mechanical ventilation in a significant percentage of patients [143]. From a molecular perspective, the glycogen accumulation interferes with cellular processes such as metabolism or autophagy. In this context, the accumulation of autophagy substrates and autophagosomes was demonstrated by using knockout mice models of GAA [144][145]. In fact, components of the autophagy system, including BECN1, GABARAP, LC3, and ATG7, are increased; however, this excess is associated with functional autophagy deficiency, as the machinery is unable to degrade them. Furthermore, in primary cells derived from KO mice, lysosomal acidification is inefficient, leading to a blockade of autophagy and an impairment of mitochondrial function, which are associated with this lysosomal disorder [146][147].
3.2. Danon Disease
Danon disease was the first LSD in which an association with autophagy involvement was reported. Danon disease it is characterized by a deficiency in LAMP2 (lysosomal-associated membrane protein 2). The disease is inherited as an X-linked trait and is exceptionally rare [148]. From a phenotypic point of view, Danon disease is characterized by severe hypertrophic cardiomyopathy, heart failure, muscle weakness, retinopathy, and different degrees of mental retardation specifically in male patients. However, in female patients, it is described as a “milder phenotype” that is mostly limited to cardiac abnormalities.
According to its relation with the autophagic process, the accumulation of large LC3-positive membrane-bound structures is a very common feature in several tissues, especially in muscular tissues. In this context, an increase in lipofuscin accumulation and myofibrillar disruption are present in cardiac muscle too.
From a molecular point of view, most Danon patients carry mutations that result in LAMP2 (lysosomal-associated membrane protein 2) loss of function. In fact, Danon disease is also known as “glycogen storage disease due to LAMP2 deficiency”. These alterations cause an important failure in autophagosomal–lysosomal fusion accompanied by an excessive accumulation of autophagosomes and partial modifications in a subset of lysosomal enzymes and p62 aggregates. In this regard, by using muscle biopsies from patients, a correlation was reported between a blockage in autophagy flux and disease severity. In addition, mice deficient in Lamp2 mimic Danon disease in humans, showing an accumulation of autophagosomes, which severely affects cardiac contractile function [149].
Although the most Danon-affected people present a deficiency of all three LAMP-2 isoforms, the pathogenesis and clinical manifestations are attributed to the specific deficiency of LAMP-2B, the expression of which is abundant in the heart, muscle, and brain. Indeed, only a defect in LAMP-2B is sufficient to cause most of the disease manifestations [150]. Furthermore, these patients present defects also in mitochondrial clearance (mitophagy), which shows that it is not only bulk autophagy that is altered in this type of disease [151] (Figure 2).
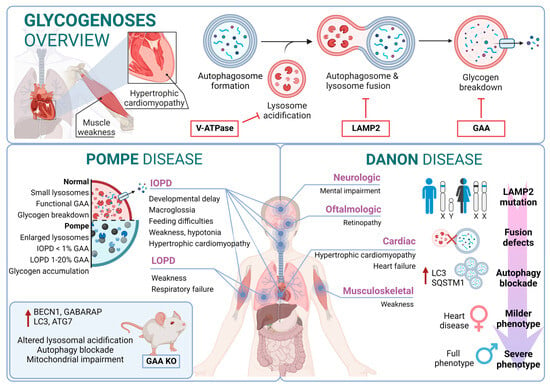
Figure 2. Glycogenoses overview. Pompe disease and Danon disease.
4. Autophagic Process in Sphingolipidoses
Sphingolipids represent a major category of lipids in the nervous system and play an important role in neural development and functionality [152]. Their metabolism is tightly regulated through a multistep degradation process that relies on several lysosomal hydrolases [153]. In this regard, sphingolipidoses represent a broad group of inherited disorders related to sphingolipid metabolism that frequently affect the nervous system. Specifically, sphingolipidoses are a subset of LSDs characterized by the accumulation of partially or completely undegraded sphingolipids. These alterations are observed mainly in the pediatric population, manifesting in neurodegeneration that leads to psychomotor retardation and myoclonus due to widespread and progressive damage to neurons. In some cases, these conditions may cause weakness and spasticity due to the involvement of white-matter tracts. The genetic changes underlying these disorders are diverse and can lead to the accumulation of substances such as sphingomyelin, glycolipids, glucocerebrosides, gangliosides, unesterified cholesterol, and sulfatide compounds, among others [154]. For these reasons, it is not surprising that several disturbances in autophagy have been reported in sphingolipidoses. For example, the addition of glycosphingolipids to cells, even by a simple method such as supplementation in a culture medium, triggers autophagy, prevents the clearance of autophagosomes, and leads to their accumulation [155]. Diseases in this category include Niemann–Pick, Gaucher, and Fabry diseases as well as mucolipidoses, and GM1/2 gangliosidoses such as Tay–Sachs and Sandhoff diseases [156]. Mutations related to sphingolipidoses are listed in Supplementary Table S3 [157][158][159][160][161][162][163][164][165][166][167][168][169][170][171][172][173][174][175][176][177].
4.1. Gaucher Disease
Gaucher disease (GD) is one of the most common lysosomal storage disorders and occurs in up to 1 in 40,000 live births in the general population. GD disease includes three clinical phenotypes: GD types I, II, and III [178]. GD type I is considered a systemic disorder, with no neurological involvement. This type represents more than 90% of GD cases; the common symptoms are splenomegaly, hepatomegaly, thrombocytopenia, coagulation abnormalities, anemia, bone disease, and bone marrow infiltration by storage cells. The age of onset is highly variable and can start at any time from childhood to 70 years old. However, most patients are diagnosed before the age of 20 [179].
Several studies pointed to a genetic association between the p.N370S allele, both homozygous and heterozygous states, and the development of GD type I [180]. In contrast, GD type II often manifests with severe symptoms within the first six months of life. Patients with this form of the disease experience neurological manifestations, brainstem involvement, developmental delays, cachexia, respiratory distress, pneumonia, skin issues, and other symptoms. The prognosis for individuals with GD type II is very poor, as infants rapidly deteriorate, and unfortunately, most do not survive beyond 2 years of age [181]. GD type III (commonly known as chronic neuropathic form) has a more insidious course. The manifestations are developmental delay; strabismus and supranuclear gaze palsy; progressive dementia; myoclonus; corneal opacities; and cardiovascular calcification. From a genetic point of view, it is known that individuals who are homozygous for the p.L444P allele probably develop type II or III [182][183]. In this regard, glucosylceramide and glucosylsphingosine are the primary glycosphingolipids that accumulate in this disease, primarily in macrophages known as “Gaucher cells”, which are found in the liver, spleen, lungs, and central nervous system. From a molecular point of view, these accumulations result from mutations in the GBA1 gene, which encodes glucocerebrosidase [184].
In GD, there is an impairment of the autophagic process. In fact, it was described as an accumulation of autophagosomes in different in vitro and in vivo models. For example, using induced pluripotent stem cells (iPSCs) derived from Gaucher patient cells that were reprogrammed into neurons, the researchers observed an increase in the number of autophagosomes and the amount of autophagic markers such as LC3-II and p62, which appear exclusively in neuropathic cells. Specifically, affected neuropathic cells display blocked autophagic flux with reduced autophagosomal clearance and decreased levels of LAMP1 and TFEB. This indicates a lysosomal dysfunction and is a probable cause of neurodegeneration in this pathology. The overexpression of TFEB in combination with recombinant glucocerebrosidase treatment ameliorates these alterations [185].
Furthermore, a mouse model that phenocopies GD mediated by mutations in both glucocerebrosidase (V394L) and C-saposin deficiencies shows accumulation of p62/SQSTM1 in neurons and astrocytes along with sequestration of undigested materials within axonal vesicles. These observations indicate that autolysosomal cargo degradation is impaired in cells affected by GD [186].
In summary, autophagy dysfunction is present in various model systems of GD, but the underlying mechanisms are still unclear.
4.2. Niemann–Pick Type C Disease (NPC)
Niemann–Pick type C (NPC) disease is a genetic autosomal recessive lysosomal storage disorder caused by mutations in either NPC1 (95% of cases) or NPC2 (5% of cases). These genes encode proteins involved in the intracellular trafficking of lipids and cholesterol [187]. Mutations in these genes result in the accumulation of unesterified cholesterol in the liver, spleen, and brain, which, in turn, disrupts lipid transport. In fact, these alterations cause a disruption that leads to the loss of Purkinje cells in the cerebellum and degeneration of other components of the central nervous system [188].
From a clinical perspective, NPC is typically a disease with juvenile or later onset, and the rate of progression inversely correlates with the age of onset. Common symptoms of NPC include ataxia, splenomegaly, hepatomegaly, hypotonia, severe liver disease, respiratory infections, and abnormal eye movements [189].
In this context, there is an alteration of the autophagic mechanism in NPC, as an accumulation of autophagosomes in skin fibroblasts from NPC patients is described. In the molecular landscape of this disease, this accumulation is partially due to the function of BECN1 and LC3B. In wild-type fibroblasts, their levels increase when exposed to U18666A, a small molecule used to induce NPC-like lipid trafficking defects. Moreover, NPC exhibits a blocked autophagic flux due to impaired autophagosome maturation [190] and specific defects in mitophagy [191].
Therefore, autophagy is significantly disrupted in NPC. This disruption interferes in the maintenance of cellular and tissue homeostasis, contributing to the pathological changes observed in NPC patients. The accumulation of autophagosomes, their impaired maturation, and the defective mitochondrial function all contribute to the disease’s progression, affecting cellular and tissue health.
4.3. Fabry Disease
Fabry disease (FD) is an X-linked LSD characterized by mutations in the GLA gene. This gene encodes the lysosomal enzyme α-galactosidase A (α-Gal A). Deficiency of α-Gal A causes an accumulation of globotriaosylceramide (Gb3) in organs, including the heart, kidneys, brain, and eyes, among others [192]. FD is among the more frequent LSDs after GD [193]. Due to its X-linked inheritance pattern, prevalence in males is higher than in females. In males, there are two subtypes: the classic presentation with a severe phenotype and the nonclassic with a less severe phenotype [194]. The classical phenotype of FD has an incidence of 1:22,000 to 1:40,000 in males, while the nonclassical form oscillates from 1:1000 to 1:3000 in males and 1:6000 to 1:40,000 in females. In this regard, patients with the classic phenotype have less than 1% of normal α-Gal A activity and tend to develop complications and symptoms earlier in life. Patients with the nonclassic subtype have a milder form of the disease, with higher α-Gal A activity [195]. In contrast, although females are typically asymptomatic, a small percentage of them can exhibit a milder pattern of the disease due to continued secretion of α-Gal A from their other X chromosome.
Clinical features of FD include auditory disturbances, renal disease, cerebrovascular disease, cardiac hypertrophy, arrhythmia, angiokeratoma (skin lesions), and excessive sweating [196]. From a molecular perspective, over 900 mutations have been identified in association with FD; D313Y, E66Q, and A143T are among the most common associated mutations [197][198].
In this scenario, taking into account the rest of the sphingolipidoses, in FD there is an increased basal expression of the autophagosome marker LC3-II, as observed in cultured cells from FD patients compared to wild-type cells. Furthermore, human podocytes derived from patients with FD have increased expression of GABARAP and BECN1 along with an inhibition of mTOR. However, an impairment in the autophagy flux in FD is described. According to this alteration, an increased staining of p62 and ubiquitin was observed in renal tissues and cultured fibroblasts from FD patients [199]. Moreover, the accumulation of autophagy substrates, autophagosomes, and lysosomes was proved using an α-Gal A-deficient mouse model [200]. These changes suggest that autophagy dysfunction could contribute to the progression of neuropathological changes in FD [201] (Figure 3).
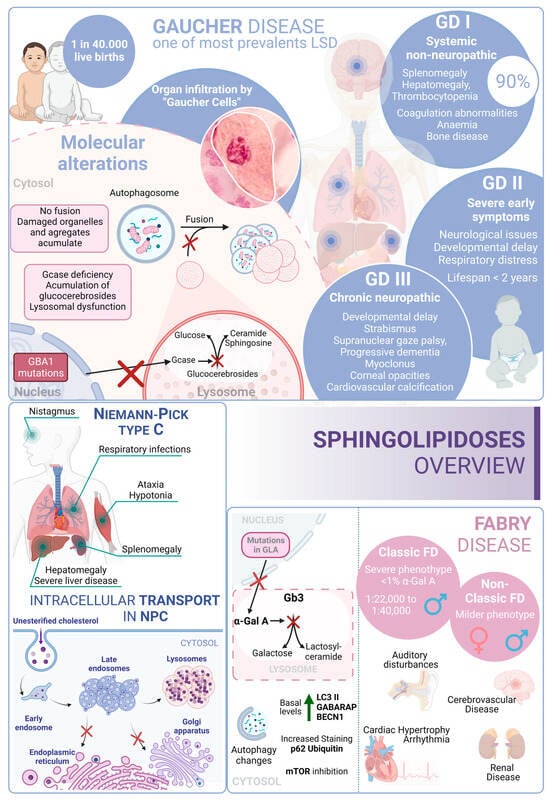
Figure 3. Sphingolipidoses overview. Gaucher disease, Niemann–Pick type C disease, and Fabry disease. GD I, II and III correspond to the three different presentations of Gaucher Disease.
5. Autophagy Pathway in Neuronal Ceroid Lipofuscinoses
Neuronal ceroid lipofuscinoses (NCLs) are one of the most prevalent causes of neurodegeneration in children. NCLs typically present with symptoms such as blindness, seizures, progressive cognitive decline, and motor impairment. NCLs exhibit both genetic and phenotypic diversity. The juvenile onset NCL (JNCL), also known as Batten disease, is the most common presentation and is caused by mutations in the CLN3 gene [202]. However, NCLs could manifest as different subtypes, each named according to the specific mutations in specific ceroid lipofuscinosis neuronal (CLN) genes such as CLN1, CLN2, CLN4, CLN5, CLN6, CLN7, CLN8, CLN10, CLN11, CLN12, or CLN13 [203]. Mutations related to NCLs are listed in Supplementary Table S4 [204][205][206][207][208][209][210][211][212][213][214][215][216][217][218][219][220][221][222][223][224][225].
The predicted functions of CLN proteins are diverse, with some acting as lysosomal enzymes, while others are thought to regulate intracellular trafficking or membrane transport. Unfortunately, the precise cellular roles of most CLN proteins remain a mystery, prompting the utilization of a wide variety of model systems in NCL research. However, these diseases share the common features of autophagosome accumulation, dysfunctional mitochondria, and alterations in autophagy-related pathways, providing important insights into their pathogenesis [226][227].
Batten disease is an autosomal recessive disorder typically affecting children between the ages of 5 and 10 caused by mutations in CLN3. A frequently observed defect in CLN3 is the homozygous deletion of 966 base pairs, encompassing exons 7 and 8, resulting in a premature stop codon in exon 9 [228]. In this context, the CLN3 protein is located in many cellular compartments, including the endo-lysosomal pathway and the Golgi complex. However, although its exact function is not fully understood, it has been linked to intracellular trafficking through interactions with Rab7A and protein secretion processes [229][230].
In this context, several mammalian models of NCLIII disease have shown reduced trafficking and levels of lysosomal enzymes. Early-stage defects in the autophagy pathway have been observed in both mouse and human cellular models with CLN3 mutations, leading to the accumulation of autophagosomes and autolysosomes. In particular, Cln3Dex7/8 knock-in mice and Cln3Dex7/8 cerebellar cells exhibit increased levels of LC3-II, downregulation of mTORC1, accumulation of autophagosomes, and impaired maturation as well as impaired turnover of ATP synthase subunit C [231][232].
On the other hand, the NCL X subtype (characterized by mutations in the Cathepsin D gene) also presents an increase in the number of autophagosomes and an accumulation of malfunctioning mitochondria due to an impairment in the autophagy flux [233] (Figure 4).
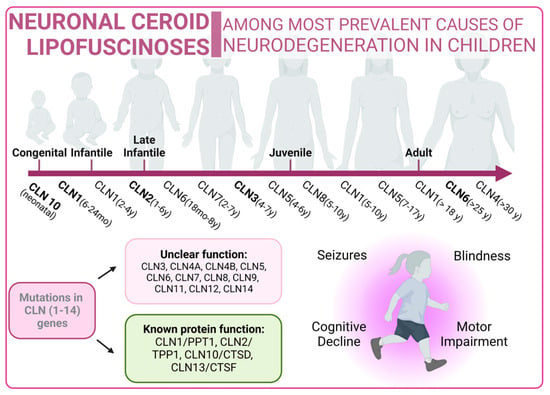
Figure 4. Neuronal Ceroid Lipofuscinoses overview. Classical forms of the disease are marked in bold type.
6. Autophagy in Glycoproteinoses
Glycoproteinoses form a category of lysosomal diseases resulting from deficiencies in the catabolism of glycoproteins. In this regard, glycoproteins are prevalent components found in cells and on cell surfaces. These genetic disorders follow an autosomal recessive inheritance pattern. Glycoproteinoses share the deficit of specific lysosomal enzymes that are crucial for the systematic breakdown of glycoprotein glucids [234]. Pathogenic sequence variants in genes encoding these enzymes lead to glycoproteinoses. The oligosaccharide composition serves as an indicator of glycoproteinosis, potentially offering insights into a specific diagnosis [235]. In this context, there are some deficiencies encompassed in this type of pathology, such as fucosidosis, galactosialidosis, Schindler’s disease, α-Mannosidosis, or β-Mannosidosis, among others [236]. In many of these disorders, the accumulation of undigested material induces vacuolization in cells, such as peripheral blood cells and fibroblasts. This accumulation can also have pleiotropic effects on cellular functions, including synaptic release, exocytosis, and autophagy [235]. Mutations related to glycoproteinoses are listed in Supplementary Table S5 [237][238][239][240][241][242][243][244][245][246][247][248][249][250][251].
6.1. α-Mannosidosis
α-mannosidosis, is an uncommon LSD (1:500.000) and follows an autosomal recessive inheritance pattern. Typically, it is unnoticeable at birth and displays symptoms progressively [252]. Clinical features of this disease include cognitive developmental delay, hearing loss, skeletal deformities, central nervous system involvement, and immunodeficiencies. Traditionally, α-mannosidosis is classified into two categories, according to its severity. However, wider classification includes three clinical types differentiated by the age of onset, speed of progression, and presence/absence of skeletal abnormalities. Type 1 includes mild presentation, an age of onset after 10 years old, no skeletal implication, and very slow progression. Type 2 is the moderate form, in which symptoms are identified before 10 years old, including skeletal abnormalities, with a slow progression that leads to ataxia around the age of 30. Type 3 is the most severe presentation and is immediately recognizable, exhibiting skeletal abnormalities. It progresses fast, culminating in early death due to central nervous system involvement or myopathy. The most frequent presentation is Type 2 [253][254][255].
From a genetic point of view, α-mannosidosis is originated by MAN2B1 gene mutations, generating an aberrant enzyme (α-mannosidase) that is unable to break down mannose-containing oligosaccharides [256]. Consequently, these accumulate within lysosomes, triggering cell malfunction and eventual cell death. The build-up of oligosaccharides and ensuing cell death contribute to tissue and organ damage, resulting in the distinctive features observed in α-mannosidosis [257].
6.2. β-Mannosidosis
β-mannosidosis is an autosomal recessive LSD that occurs as a result of a dysfunctional β-mannosidase enzyme. While well-known and relatively common in goats and other types of cattle, it is exceptionally rare in humans. For this reason, the characterization of this pathology in a human context poses significant challenges due to its infrequency and limited occurrence in the human population [258][259].
The signs and symptoms of β-mannosidosis present a wide spectrum of severity, and onset can occur from infancy to adulthood. In fact, there is no clear genotype/phenotype correlation. Common symptoms include intellectual disability, and some may experience delayed motor development and seizures. Behavioral problems, such as hyperactivity, impulsivity, aggressiveness, or a tendency to depression; respiratory and ear infections; hearing loss; speech impairment; swallowing difficulties; and hypotonia, are additional challenges faced by those with β-mannosidosis and are also common among patients who may exhibit introverted behavior [260][261]. Distinctive facial features and the presence of small angiokeratomas, formed by clusters of enlarged blood vessels, are also observed in individuals with this condition.
From a molecular point of view, β-mannosidosis is caused by alterations in the MANBA gene [262]. These disrupt the function of the β-mannosidase enzyme, which serves as the final exoglycosidase in the degradation of N-linked oligosaccharides of glycoproteins, removing β-linked mannose residues [263]. Affected individuals exhibit a significant reduction in β-mannosidase activity, and this leads to the lysosomal accumulation of disaccharides.
References
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486.
- Palhegyi, A.M.; Seranova, E.; Dimova, S.; Hoque, S.; Sarkar, S. Biomedical Implications of Autophagy in Macromolecule Storage Disorders. Front. Cell Dev. Biol. 2019, 7, 179.
- Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of Mucopolysaccharidoses, an Update. Int. J. Mol. Sci. 2020, 21, 2515.
- Gaffke, L.; Pierzynowska, K.; Podlacha, M.; Brokowska, J.; Wegrzyn, G. Changes in cellular processes occurring in mucopolysaccharidoses as underestimated pathomechanisms of these diseases. Cell Biol. Int. 2021, 45, 498–506.
- Sly, W.S.; Vogler, C.; Grubb, J.H.; Zhou, M.; Jiang, J.; Zhou, X.Y.; Tomatsu, S.; Bi, Y.; Snella, E.M. Active site mutant transgene confers tolerance to human β-glucuronidase without affecting the phenotype of MPS VII mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2205–2210.
- Gabrielli, O.; Coppa, G.V.; Bruni, S.; Villani, G.R.; Pontarelli, G.; Di Natale, P. An adult Sanfilippo type A patient with homozygous mutation R206P in the sulfamidase gene. Am. J. Med. Genet. Part A 2005, 133, 85–89.
- Tetreault, M.; Gonzalez, M.; Dicaire, M.J.; Allard, P.; Gehring, K.; Leblanc, D.; Leclerc, N.; Schondorf, R.; Mathieu, J.; Zuchner, S.; et al. Adult-onset painful axonal polyneuropathy caused by a dominant NAGLU mutation. Brain J. Neurol. 2015, 138, 1477–1483.
- Di Natale, P.; Villani, G.R.; Di Domenico, C.; Daniele, A.; Dionisi Vici, C.; Bartuli, A. Analysis of Sanfilippo A gene mutations in a large pedigree. Clin. Genet. 2003, 63, 314–318.
- Takai, T.; Higaki, K.; Aguilar-Moncayo, M.; Mena-Barragan, T.; Hirano, Y.; Yura, K.; Yu, L.; Ninomiya, H.; Garcia-Moreno, M.I.; Sakakibara, Y.; et al. A bicyclic 1-deoxygalactonojirimycin derivative as a novel pharmacological chaperone for GM1 gangliosidosis. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 526–532.
- Ishii, N.; Oohira, T.; Oshima, A.; Sakuraba, H.; Endo, F.; Matsuda, I.; Sukegawa, K.; Orii, T.; Suzuki, Y. Clinical and molecular analysis of a Japanese boy with Morquio B disease. Clin. Genet. 1995, 48, 103–108.
- Chiong, M.A.; Canson, D.M.; Abacan, M.A.; Baluyot, M.M.; Cordero, C.P.; Silao, C.L. Clinical, biochemical and molecular characteristics of Filipino patients with mucopolysaccharidosis type II—Hunter syndrome. Orphanet J. Rare Dis. 2017, 12, 7.
- Yassaee, V.R.; Hashemi-Gorji, F.; Miryounesi, M.; Rezayi, A.; Ravesh, Z.; Yassaee, F.; Salehpour, S. Clinical, biochemical and molecular features of Iranian families with mucopolysaccharidosis: A case series. Clin. Chim. Acta Int. J. Clin. Chem. 2017, 474, 88–95.
- Tylki-Szymanska, A.; Czartoryska, B.; Bunge, S.; van Diggelen, O.P.; Kleijer, W.J.; Poorthuis, B.J.; Huijmans, J.G.; Gorska, D. Clinical, biochemical and molecular findings in a two-generation Morquio A family. Clin. Genet. 1998, 53, 369–374.
- Flomen, R.H.; Green, P.M.; Bentley, D.R.; Giannelli, F.; Green, E.P. Detection of point mutations and a gross deletion in six Hunter syndrome patients. Genomics 1992, 13, 543–550.
- Bonuccelli, G.; Di Natale, P.; Corsolini, F.; Villani, G.; Regis, S.; Filocamo, M. The effect of four mutations on the expression of iduronate-2-sulfatase in mucopolysaccharidosis type II. Biochim. Biophys. Acta 2001, 1537, 233–238.
- Santamaria, R.; Chabas, A.; Callahan, J.W.; Grinberg, D.; Vilageliu, L. Expression and characterization of 14 GLB1 mutant alleles found in GM1-gangliosidosis and Morquio B patients. J. Lipid Res. 2007, 48, 2275–2282.
- Montfort, M.; Garrido, E.; Hopwood, J.J.; Grinberg, D.; Chabas, A.; Vilageliu, L. Expression and functional characterization of human mutant sulfamidase in insect cells. Mol. Genet. Metab. 2004, 83, 246–251.
- Iwasaki, H.; Watanabe, H.; Iida, M.; Ogawa, S.; Tabe, M.; Higaki, K.; Nanba, E.; Suzuki, Y. Fibroblast screening for chaperone therapy in β-galactosidosis. Brain Dev. 2006, 28, 482–486.
- Yamada, S.; Tomatsu, S.; Sly, W.S.; Islam, R.; Wenger, D.A.; Fukuda, S.; Sukegawa, K.; Orii, T. Four novel mutations in mucopolysaccharidosis type VII including a unique base substitution in exon 10 of the β-glucuronidase gene that creates a novel 5′-splice site. Hum. Mol. Genet. 1995, 4, 651–655.
- Tieu, P.T.; Bach, G.; Matynia, A.; Hwang, M.; Neufeld, E.F. Four novel mutations underlying mild or intermediate forms of α-L-iduronidase deficiency (MPS IS and MPS IH/S). Hum. Mutat. 1995, 6, 55–59.
- Mok, A.; Cao, H.; Hegele, R.A. Genomic basis of mucopolysaccharidosis type IIID (MIM 252940) revealed by sequencing of GNS encoding N-acetylglucosamine-6-sulfatase. Genomics 2003, 81, 1–5.
- Shi, L.; Li, B.; Huang, Y.; Ling, X.; Liu, T.; Lyon, G.J.; Xu, A.; Wang, K. “Genotype-first” approaches on a curious case of idiopathic progressive cognitive decline. BMC Med. Genom. 2014, 7, 66.
- Pineda, T.; Marie, S.; Gonzalez, J.; Garcia, A.L.; Acosta, A.; Morales, M.; Correa, L.N.; Vivas, R.; Escobar, X.; Protzel, A.; et al. Genotypic and bioinformatic evaluation of the α-l-iduronidase gene and protein in patients with mucopolysaccharidosis type I from Colombia, Ecuador and Peru. Mol. Genet. Metab. Rep. 2014, 1, 468–473.
- Brunetti-Pierri, N.; Scaglia, F. GM1 gangliosidosis: Review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metab. 2008, 94, 391–396.
- Gaffke, L.; Pierzynowska, K.; Piotrowska, E.; Wegrzyn, G. How close are we to therapies for Sanfilippo disease? Metab. Brain Dis. 2018, 33, 1–10.
- Oshima, A.; Yoshida, K.; Shimmoto, M.; Fukuhara, Y.; Sakuraba, H.; Suzuki, Y. Human β-galactosidase gene mutations in morquio B disease. Am. J. Hum. Genet. 1991, 49, 1091–1093.
- Teng, Y.N.; Wang, T.R.; Hwu, W.L.; Lin, S.P.; Lee-Chen, G.J. Identification and characterization of -3c-g acceptor splice site mutation in human α-L-iduronidase associated with mucopolysaccharidosis type IH/S. Clin. Genet. 2000, 57, 131–136.
- Bunge, S.; Ince, H.; Steglich, C.; Kleijer, W.J.; Beck, M.; Zaremba, J.; van Diggelen, O.P.; Weber, B.; Hopwood, J.J.; Gal, A. Identification of 16 sulfamidase gene mutations including the common R74C in patients with mucopolysaccharidosis type IIIA (Sanfilippo A). Hum. Mutat. 1997, 10, 479–485.
- Bunge, S.; Kleijer, W.J.; Tylki-Szymanska, A.; Steglich, C.; Beck, M.; Tomatsu, S.; Fukuda, S.; Poorthuis, B.J.; Czartoryska, B.; Orii, T.; et al. Identification of 31 novel mutations in the N-acetylgalactosamine-6-sulfatase gene reveals excessive allelic heterogeneity among patients with Morquio A syndrome. Hum. Mutat. 1997, 10, 223–232.
- Weber, B.; van de Kamp, J.J.; Kleijer, W.J.; Guo, X.H.; Blanch, L.; van Diggelen, O.P.; Wevers, R.; Poorthuis, B.J.; Hopwood, J.J. Identification of a common mutation (R245H) in Sanfilippo A patients from The Netherlands. J. Inherit. Metab. Dis. 1998, 21, 416–422.
- Scott, H.S.; Litjens, T.; Nelson, P.V.; Thompson, P.R.; Brooks, D.A.; Hopwood, J.J.; Morris, C.P. Identification of mutations in the α-L-iduronidase gene (IDUA) that cause Hurler and Scheie syndromes. Am. J. Hum. Genet. 1993, 53, 973–986.
- Fan, X.; Zhang, H.; Zhang, S.; Bagshaw, R.D.; Tropak, M.B.; Callahan, J.W.; Mahuran, D.J. Identification of the gene encoding the enzyme deficient in mucopolysaccharidosis IIIC (Sanfilippo disease type C). Am. J. Hum. Genet. 2006, 79, 738–744.
- Garrido, E.; Chabas, A.; Coll, M.J.; Blanco, M.; Dominguez, C.; Grinberg, D.; Vilageliu, L.; Cormand, B. Identification of the molecular defects in Spanish and Argentinian mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) patients, including 9 novel mutations. Mol. Genet. Metab. 2007, 92, 122–130.
- Litjens, T.; Brooks, D.A.; Peters, C.; Gibson, G.J.; Hopwood, J.J. Identification, expression, and biochemical characterization of N-acetylgalactosamine-4-sulfatase mutations and relationship with clinical phenotype in MPS-VI patients. Am. J. Hum. Genet. 1996, 58, 1127–1134.
- Harmatz, P.R.; Mengel, E.; Geberhiwot, T.; Muschol, N.; Hendriksz, C.J.; Burton, B.K.; Jameson, E.; Berger, K.I.; Jester, A.; Treadwell, M.; et al. Impact of elosulfase alfa in patients with morquio A syndrome who have limited ambulation: An open-label, phase 2 study. Am. J. Med. Genet. Part A 2017, 173, 375–383.
- Heron, B.; Mikaeloff, Y.; Froissart, R.; Caridade, G.; Maire, I.; Caillaud, C.; Levade, T.; Chabrol, B.; Feillet, F.; Ogier, H.; et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am. J. Med. Genet. Part A 2011, 155, 58–68.
- Vervoort, R.; Gitzelmann, R.; Bosshard, N.; Maire, I.; Liebaers, I.; Lissens, W. Low β-glucuronidase enzyme activity and mutations in the human β-glucuronidase gene in mild mucopolysaccharidosis type VII, pseudodeficiency and a heterozygote. Hum. Genet. 1998, 102, 69–78.
- Coutinho, M.F.; Lacerda, L.; Macedo-Ribeiro, S.; Baptista, E.; Ribeiro, H.; Prata, M.J.; Alves, S. Lysosomal multienzymatic complex-related diseases: A genetic study among Portuguese patients. Clin. Genet. 2012, 81, 379–393.
- Vervoort, R.; Lissens, W.; Liebaers, I. Molecular analysis of a patient with hydrops fetalis caused by β-glucuronidase deficiency, and evidence for additional pseudogenes. Hum. Mutat. 1993, 2, 443–445.
- Bach, G.; Moskowitz, S.M.; Tieu, P.T.; Matynia, A.; Neufeld, E.F. Molecular analysis of Hurler syndrome in Druze and Muslim Arab patients in Israel: Multiple allelic mutations of the IDUA gene in a small geographic area. Am. J. Hum. Genet. 1993, 53, 330–338.
- Mangas, M.; Nogueira, C.; Prata, M.J.; Lacerda, L.; Coll, M.J.; Soares, G.; Ribeiro, G.; Amaral, O.; Ferreira, C.; Alves, C.; et al. Molecular analysis of mucopolysaccharidosis type IIIB in Portugal: Evidence of a single origin for a common mutation (R234C) in the Iberian Peninsula. Clin. Genet. 2008, 73, 251–256.
- Canals, I.; Elalaoui, S.C.; Pineda, M.; Delgadillo, V.; Szlago, M.; Jaouad, I.C.; Sefiani, A.; Chabas, A.; Coll, M.J.; Grinberg, D.; et al. Molecular analysis of Sanfilippo syndrome type C in Spain: Seven novel HGSNAT mutations and characterization of the mutant alleles. Clin. Genet. 2011, 80, 367–374.
- Tanaka, A.; Kimura, M.; Lan, H.T.; Takaura, N.; Yamano, T. Molecular analysis of the α-N-acetylglucosaminidase gene in seven Japanese patients from six unrelated families with mucopolysaccharidosis IIIB (Sanfilippo type B), including two novel mutations. J. Hum. Genet. 2002, 47, 484–487.
- Li, P.; Bellows, A.B.; Thompson, J.N. Molecular basis of iduronate-2-sulphatase gene mutations in patients with mucopolysaccharidosis type II (Hunter syndrome). J. Med. Genet. 1999, 36, 21–27.
- Zhao, H.G.; Li, H.H.; Bach, G.; Schmidtchen, A.; Neufeld, E.F. The molecular basis of Sanfilippo syndrome type B. Proc. Natl. Acad. Sci. USA 1996, 93, 6101–6105.
- Chistiakov, D.A.; Savost’anov, K.V.; Kuzenkova, L.M.; Gevorkyan, A.K.; Pushkov, A.A.; Nikitin, A.G.; Pakhomov, A.V.; Vashakmadze, N.D.; Zhurkova, N.V.; Podkletnova, T.V.; et al. Molecular characteristics of patients with glycosaminoglycan storage disorders in Russia. Clin. Chim. Acta; Int. J. Clin. Chem. 2014, 436, 112–120.
- Pollard, L.M.; Jones, J.R.; Wood, T.C. Molecular characterization of 355 mucopolysaccharidosis patients reveals 104 novel mutations. J. Inherit. Metab. Dis. 2013, 36, 179–187.
- Giraldo, G.A.; Ayala-Ramirez, P.; Prieto, J.C.; Garcia-Robles, R.; Acosta, J.C. Molecular findings of Colombian patients with type VI mucopolysaccharidosis (Maroteaux-Lamy syndrome). Meta Gene 2016, 7, 83–89.
- Aronovich, E.L.; Pan, D.; Whitley, C.B. Molecular genetic defect underlying α-L-iduronidase pseudodeficiency. Am. J. Hum. Genet. 1996, 58, 75–85.
- Wang, Z.; Zhang, W.; Wang, Y.; Meng, Y.; Su, L.; Shi, H.; Huang, S. Mucopolysaccharidosis IVA mutations in Chinese patients: 16 novel mutations. J. Hum. Genet. 2010, 55, 534–540.
- Montano, A.M.; Kaitila, I.; Sukegawa, K.; Tomatsu, S.; Kato, Z.; Nakamura, H.; Fukuda, S.; Orii, T.; Kondo, N. Mucopolysaccharidosis IVA: Characterization of a common mutation found in Finnish patients with attenuated phenotype. Hum. Genet. 2003, 113, 162–169.
- Clarke, L.A. Mucopolysaccharidosis Type I. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Lee-Chen, G.J.; Lin, S.P.; Tang, Y.F.; Chin, Y.W. Mucopolysaccharidosis type I: Characterization of novel mutations affecting α-L-iduronidase activity. Clin. Genet. 1999, 56, 66–70.
- Bunge, S.; Kleijer, W.J.; Steglich, C.; Beck, M.; Zuther, C.; Morris, C.P.; Schwinger, E.; Hopwood, J.J.; Scott, H.S.; Gal, A. Mucopolysaccharidosis type I: Identification of 8 novel mutations and determination of the frequency of the two common α-L-iduronidase mutations (W402X and Q70X) among European patients. Hum. Mol. Genet. 1994, 3, 861–866.
- Lee-Chen, G.J.; Wang, T.R. Mucopolysaccharidosis type I: Identification of novel mutations that cause Hurler/Scheie syndrome in Chinese families. J. Med. Genet. 1997, 34, 939–941.
- Valstar, M.J.; Neijs, S.; Bruggenwirth, H.T.; Olmer, R.; Ruijter, G.J.; Wevers, R.A.; van Diggelen, O.P.; Poorthuis, B.J.; Halley, D.J.; Wijburg, F.A. Mucopolysaccharidosis type IIIA: Clinical spectrum and genotype-phenotype correlations. Ann. Neurol. 2010, 68, 876–887.
- Tang, J.; Pan, J.; Guo, Y.; Ai, Y.; Jiang, W.; Du, M.; Fang, Q. Mucopolysaccharidosis type IIIB mutations in Chinese patients: Identification of two novel NAGLU mutations and analysis of two cases involving prenatal diagnosis. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 419, 33–38.
- Yogalingam, G.; Weber, B.; Meehan, J.; Rogers, J.; Hopwood, J.J. Mucopolysaccharidosis type IIIB: Characterisation and expression of wild-type and mutant recombinant α-N-acetylglucosaminidase and relationship with sanfilippo phenotype in an attenuated patient. Biochim. Biophys. Acta 2000, 1502, 415–425.
- Fukuda, S.; Tomatsu, S.; Masue, M.; Sukegawa, K.; Iwata, H.; Ogawa, T.; Nakashima, Y.; Hori, T.; Yamagishi, A.; Hanyu, Y. Mucopolysaccharidosis type IVA. N-acetylgalactosamine-6-sulfate sulfatase exonic point mutations in classical Morquio and mild cases. J. Clin. Investig. 1992, 90, 1049–1053.
- Jin, W.D.; Jackson, C.E.; Desnick, R.J.; Schuchman, E.H. Mucopolysaccharidosis type VI: Identification of three mutations in the arylsulfatase B gene of patients with the severe and mild phenotypes provides molecular evidence for genetic heterogeneity. Am. J. Hum. Genet. 1992, 50, 795–800.
- Tomatsu, S.; Fukuda, S.; Sukegawa, K.; Ikedo, Y.; Yamada, S.; Yamada, Y.; Sasaki, T.; Okamoto, H.; Kuwahara, T.; Yamaguchi, S.; et al. Mucopolysaccharidosis type VII: Characterization of mutations and molecular heterogeneity. Am. J. Hum. Genet. 1991, 48, 89–96.
- Wicker, G.; Prill, V.; Brooks, D.; Gibson, G.; Hopwood, J.; von Figura, K.; Peters, C. Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). An intermediate clinical phenotype caused by substitution of valine for glycine at position 137 of arylsulfatase B. J. Biol. Chem. 1991, 266, 21386–21391.
- Montfort, M.; Vilageliu, L.; Garcia-Giralt, N.; Guidi, S.; Coll, M.J.; Chabas, A.; Grinberg, D. Mutation 1091delC is highly prevalent in Spanish Sanfilippo syndrome type A patients. Hum. Mutat. 1998, 12, 274–279.
- Isogai, K.; Sukegawa, K.; Tomatsu, S.; Fukao, T.; Song, X.Q.; Yamada, Y.; Fukuda, S.; Orii, T.; Kondo, N. Mutation analysis in the iduronate-2-sulphatase gene in 43 Japanese patients with mucopolysaccharidosis type II (Hunter disease). J. Inherit. Metab. Dis. 1998, 21, 60–70.
- Bunge, S.; Steglich, C.; Beck, M.; Rosenkranz, W.; Schwinger, E.; Hopwood, J.J.; Gal, A. Mutation analysis of the iduronate-2-sulfatase gene in patients with mucopolysaccharidosis type II (Hunter syndrome). Hum. Mol. Genet. 1992, 1, 335–339.
- Tomatsu, S.; Montano, A.M.; Nishioka, T.; Gutierrez, M.A.; Pena, O.M.; Tranda Firescu, G.G.; Lopez, P.; Yamaguchi, S.; Noguchi, A.; Orii, T. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum. Mutat. 2005, 26, 500–512.
- Storch, S.; Wittenstein, B.; Islam, R.; Ullrich, K.; Sly, W.S.; Braulke, T. Mutational analysis in longest known survivor of mucopolysaccharidosis type VII. Hum. Genet. 2003, 112, 190–194.
- Shipley, J.M.; Klinkenberg, M.; Wu, B.M.; Bachinsky, D.R.; Grubb, J.H.; Sly, W.S. Mutational analysis of a patient with mucopolysaccharidosis type VII, and identification of pseudogenes. Am. J. Hum. Genet. 1993, 52, 517–526.
- Wu, B.M.; Sly, W.S. Mutational studies in a patient with the hydrops fetalis form of mucopolysaccharidosis type VII. Hum. Mutat. 1993, 2, 446–457.
- Gatti, R.; DiNatale, P.; Villani, G.R.; Filocamo, M.; Muller, V.; Guo, X.H.; Nelson, P.V.; Scott, H.S.; Hopwood, J.J. Mutations among Italian mucopolysaccharidosis type I patients. J. Inherit. Metab. Dis. 1997, 20, 803–806.
- Tomatsu, S.; Montano, A.M.; Dung, V.C.; Grubb, J.H.; Sly, W.S. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome). Hum. Mutat. 2009, 30, 511–519.
- Triggs-Raine, B.; Salo, T.J.; Zhang, H.; Wicklow, B.A.; Natowicz, M.R. Mutations in HYAL1, a member of a tandemly distributed multigene family encoding disparate hyaluronidase activities, cause a newly described lysosomal disorder, mucopolysaccharidosis IX. Proc. Natl. Acad. Sci. USA 1999, 96, 6296–6300.
- Lukong, K.E.; Landry, K.; Elsliger, M.A.; Chang, Y.; Lefrancois, S.; Morales, C.R.; Pshezhetsky, A.V. Mutations in sialidosis impair sialidase binding to the lysosomal multienzyme complex. J. Biol. Chem. 2001, 276, 17286–17290.
- Hrebicek, M.; Mrazova, L.; Seyrantepe, V.; Durand, S.; Roslin, N.M.; Noskova, L.; Hartmannova, H.; Ivanek, R.; Cizkova, A.; Poupetova, H.; et al. Mutations in TMEM76* cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Am. J. Hum. Genet. 2006, 79, 807–819.
- Litjens, T.; Morris, C.P.; Robertson, E.F.; Peters, C.; von Figura, K.; Hopwood, J.J. An N-acetylgalactosamine-4-sulfatase mutation (ΔG238) results in a severe Maroteaux-Lamy phenotype. Hum. Mutat. 1992, 1, 397–402.
- Ahmed, A.; Whitley, C.B.; Cooksley, R.; Rudser, K.; Cagle, S.; Ali, N.; Delaney, K.; Yund, B.; Shapiro, E. Neurocognitive and neuropsychiatric phenotypes associated with the mutation L238Q of the α-L-iduronidase gene in Hurler-Scheie syndrome. Mol. Genet. Metab. 2014, 111, 123–127.
- Haer-Wigman, L.; Newman, H.; Leibu, R.; Bax, N.M.; Baris, H.N.; Rizel, L.; Banin, E.; Massarweh, A.; Roosing, S.; Lefeber, D.J.; et al. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-α-glucosaminide N-acetyltransferase (HGSNAT). Hum. Mol. Genet. 2015, 24, 3742–3751.
- Elcioglu, N.H.; Pawlik, B.; Colak, B.; Beck, M.; Wollnik, B. A novel loss-of-function mutation in the GNS gene causes Sanfilippo syndrome type D. Genet. Couns. 2009, 20, 133–139.
- Seyedhassani, S.M.; Hashemi-Gorji, F.; Yavari, M.; Mirfakhraie, R. Novel missense mutation in the GALNS gene in an affected patient with severe form of mucopolysaccharidosis type IVA. Clin. Chim. Acta Int. J. Clin. Chem. 2015, 450, 121–124.
- Bhattacharya, K.; Balasubramaniam, S.; Choy, Y.S.; Fietz, M.; Fu, A.; Jin, D.K.; Kim, O.H.; Kosuga, M.; Kwun, Y.H.; Inwood, A.; et al. Overcoming the barriers to diagnosis of Morquio A syndrome. Orphanet J. Rare Dis. 2014, 9, 192.
- Shapiro, E.G.; Nestrasil, I.; Delaney, K.A.; Rudser, K.; Kovac, V.; Nair, N.; Richard, C.W., 3rd; Haslett, P.; Whitley, C.B. A Prospective Natural History Study of Mucopolysaccharidosis Type IIIA. J. Pediatr. 2016, 170, 278–287.
- Feldhammer, M.; Durand, S.; Pshezhetsky, A.V. Protein misfolding as an underlying molecular defect in mucopolysaccharidosis III type C. PLoS ONE 2009, 4, e7434.
- Bidchol, A.M.; Dalal, A.; Trivedi, R.; Shukla, A.; Nampoothiri, S.; Sankar, V.H.; Danda, S.; Gupta, N.; Kabra, M.; Hebbar, S.A.; et al. Recurrent and novel GLB1 mutations in India. Gene 2015, 567, 173–181.
- Meijer, O.L.M.; Welling, L.; Valstar, M.J.; Hoefsloot, L.H.; Bruggenwirth, H.T.; van der Ploeg, A.T.; Ruijter, G.J.G.; Wagemans, T.; Wijburg, F.A.; van Vlies, N. Residual N-acetyl-α-glucosaminidase activity in fibroblasts correlates with disease severity in patients with mucopolysaccharidosis type IIIB. J. Inherit. Metab. Dis. 2016, 39, 437–445.
- Beesley, C.; Moraitou, M.; Winchester, B.; Schulpis, K.; Dimitriou, E.; Michelakakis, H. Sanfilippo B syndrome: Molecular defects in Greek patients. Clin. Genet. 2004, 65, 143–149.
- Feldhammer, M.; Durand, S.; Mrazova, L.; Boucher, R.M.; Laframboise, R.; Steinfeld, R.; Wraith, J.E.; Michelakakis, H.; van Diggelen, O.P.; Hrebicek, M.; et al. Sanfilippo syndrome type C: Mutation spectrum in the heparan sulfate acetyl-CoA: α-glucosaminide N-acetyltransferase (HGSNAT) gene. Hum. Mutat. 2009, 30, 918–925.
- Beesley, C.E.; Burke, D.; Jackson, M.; Vellodi, A.; Winchester, B.G.; Young, E.P. Sanfilippo syndrome type D: Identification of the first mutation in the N-acetylglucosamine-6-sulphatase gene. J. Med. Genet. 2003, 40, 192–194.
- Jansen, A.C.; Cao, H.; Kaplan, P.; Silver, K.; Leonard, G.; De Meirleir, L.; Lissens, W.; Liebaers, I.; Veilleux, M.; Andermann, F.; et al. Sanfilippo syndrome type D: Natural history and identification of 3 novel mutations in the GNS Gene. Arch. Neurol. 2007, 64, 1629–1634.
- Weber, B.; Guo, X.H.; Kleijer, W.J.; van de Kamp, J.J.; Poorthuis, B.J.; Hopwood, J.J. Sanfilippo type B syndrome (mucopolysaccharidosis III B): Allelic heterogeneity corresponds to the wide spectrum of clinical phenotypes. Eur. J. Hum. Genet. EJHG 1999, 7, 34–44.
- Silva, C.M.; Severini, M.H.; Sopelsa, A.; Coelho, J.C.; Zaha, A.; d’Azzo, A.; Giugliani, R. Six novel β-galactosidase gene mutations in Brazilian patients with GM1-gangliosidosis. Hum. Mutat. 1999, 13, 401–409.
- Santamaria, R.; Chabas, A.; Coll, M.J.; Miranda, C.S.; Vilageliu, L.; Grinberg, D. Twenty-one novel mutations in the GLB1 gene identified in a large group of GM1-gangliosidosis and Morquio B patients: Possible common origin for the prevalent p.R59H mutation among gypsies. Hum. Mutat. 2006, 27, 1060.
- Alcantara-Ortigoza, M.A.; Garcia-de Teresa, B.; Gonzalez-Del Angel, A.; Berumen, J.; Guardado-Estrada, M.; Fernandez-Hernandez, L.; Navarrete-Martinez, J.I.; Maza-Morales, M.; Rius-Dominguez, R. Wide allelic heterogeneity with predominance of large IDS gene complex rearrangements in a sample of Mexican patients with Hunter syndrome. Clin. Genet. 2016, 89, 574–583.
- Pierzynowska, K.; Gaffke, L.; Jankowska, E.; Rintz, E.; Witkowska, J.; Wieczerzak, E.; Podlacha, M.; Wegrzyn, G. Proteasome Composition and Activity Changes in Cultured Fibroblasts Derived From Mucopolysaccharidoses Patients and Their Modulation by Genistein. Front. Cell Dev. Biol. 2020, 8, 540726.
- Hampe, C.S.; Yund, B.D.; Orchard, P.J.; Lund, T.C.; Wesley, J.; McIvor, R.S. Differences in MPS I and MPS II Disease Manifestations. Int. J. Mol. Sci. 2021, 22, 7888.
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129.
- De Pasquale, V.; Costanzo, M.; Siciliano, R.A.; Mazzeo, M.F.; Pistorio, V.; Bianchi, L.; Marchese, E.; Ruoppolo, M.; Pavone, L.M.; Caterino, M. Proteomic Analysis of Mucopolysaccharidosis IIIB Mouse Brain. Biomolecules 2020, 10, 355.
- Almeciga-Diaz, C.J.; Hidalgo, O.A.; Olarte-Avellaneda, S.; Rodriguez-Lopez, A.; Guzman, E.; Garzon, R.; Pimentel-Vera, L.N.; Puentes-Tellez, M.A.; Rojas-Rodriguez, A.F.; Gorshkov, K.; et al. Identification of Ezetimibe and Pranlukast as Pharmacological Chaperones for the Treatment of the Rare Disease Mucopolysaccharidosis Type IVA. J. Med. Chem. 2019, 62, 6175–6189.
- Tessitore, A.; Faella, A.; O’Malley, T.; Cotugno, G.; Doria, M.; Kunieda, T.; Matarese, G.; Haskins, M.; Auricchio, A. Biochemical, pathological, and skeletal improvement of mucopolysaccharidosis VI after gene transfer to liver but not to muscle. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 30–37.
- Bartolomeo, R.; Cinque, L.; De Leonibus, C.; Forrester, A.; Salzano, A.C.; Monfregola, J.; De Gennaro, E.; Nusco, E.; Azario, I.; Lanzara, C.; et al. mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J. Clin. Investig. 2017, 127, 3717–3729.
- Viana, G.M.; do Nascimento, C.C.; Paredes-Gamero, E.J.; D’Almeida, V. Altered Cellular Homeostasis in Murine MPS I Fibroblasts: Evidence of Cell-Specific Physiopathology. JIMD Rep. 2017, 36, 109–116.
- Vitry, S.; Bruyere, J.; Hocquemiller, M.; Bigou, S.; Ausseil, J.; Colle, M.A.; Prevost, M.C.; Heard, J.M. Storage vesicles in neurons are related to Golgi complex alterations in mucopolysaccharidosis IIIB. Am. J. Pathol. 2010, 177, 2984–2999.
- Woloszynek, J.C.; Kovacs, A.; Ohlemiller, K.K.; Roberts, M.; Sands, M.S. Metabolic adaptations to interrupted glycosaminoglycan recycling. J. Biol. Chem. 2009, 284, 29684–29691.
- Swaroop, M.; Brooks, M.J.; Gieser, L.; Swaroop, A.; Zheng, W. Patient iPSC-derived neural stem cells exhibit phenotypes in concordance with the clinical severity of mucopolysaccharidosis I. Hum. Mol. Genet. 2018, 27, 3612–3626.
- Pshezhetsky, A.V. Lysosomal storage of heparan sulfate causes mitochondrial defects, altered autophagy, and neuronal death in the mouse model of mucopolysaccharidosis III type C. Autophagy 2016, 12, 1059–1060.
- Kondo, H.; Maksimova, N.; Otomo, T.; Kato, H.; Imai, A.; Asano, Y.; Kobayashi, K.; Nojima, S.; Nakaya, A.; Hamada, Y.; et al. Mutation in VPS33A affects metabolism of glycosaminoglycans: A new type of mucopolysaccharidosis with severe systemic symptoms. Hum. Mol. Genet. 2017, 26, 173–183.
- Malicdan, M.C.; Nishino, I. Autophagy in lysosomal myopathies. Brain Pathol. 2012, 22, 82–88.
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749.
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779.
- Tsunoda, H.; Ohshima, T.; Tohyama, J.; Sasaki, M.; Sakuragawa, N.; Martiniuk, F. Acid α-glucosidase deficiency: Identification and expression of a missense mutation (S529V) in a Japanese adult phenotype. Hum. Genet. 1996, 97, 496–499.
- Angelini, C.; Nascimbeni, A.C.; Fanin, M. Autophagy in Natural History and After ERT in Glycogenosis Type II. JIMD Rep. 2015, 21, 71–77.
- Chen, X.; Liu, T.; Huang, M.; Wu, J.; Zhu, J.; Guo, Y.; Xu, X.; Li, F.; Wang, J.; Fu, L. Clinical and Molecular Characterization of Infantile-Onset Pompe Disease in Mainland Chinese Patients: Identification of Two Common Mutations. Genet. Test. Mol. Biomark. 2017, 21, 391–396.
- Emilsson, V.; Ilkov, M.; Lamb, J.R.; Finkel, N.; Gudmundsson, E.F.; Pitts, R.; Hoover, H.; Gudmundsdottir, V.; Horman, S.R.; Aspelund, T.; et al. Co-regulatory networks of human serum proteins link genetics to disease. Science 2018, 361, 769–773.
- Di Blasi, C.; Jarre, L.; Blasevich, F.; Dassi, P.; Mora, M. Danon disease: A novel LAMP2 mutation affecting the pre-mRNA splicing and causing aberrant transcripts and partial protein expression. Neuromuscul. Disord. NMD 2008, 18, 962–966.
- Cheng, Z.; Fang, Q. Danon disease: Focusing on heart. J. Hum. Genet. 2012, 57, 407–410.
- Charron, P.; Villard, E.; Sebillon, P.; Laforet, P.; Maisonobe, T.; Duboscq-Bidot, L.; Romero, N.; Drouin-Garraud, V.; Frebourg, T.; Richard, P.; et al. Danon’s disease as a cause of hypertrophic cardiomyopathy: A systematic survey. Heart 2004, 90, 842–846.
- Papadopoulos, C.; Orlikowski, D.; Prigent, H.; Lacour, A.; Tard, C.; Furby, A.; Praline, J.; Sole, G.; Hogrel, J.Y.; De Antonio, M.; et al. Effect of enzyme replacement therapy with alglucosidase alfa (Myozyme®) in 12 patients with advanced late-onset Pompe disease. Mol. Genet. Metab. 2017, 122, 80–85.
- Shin, Y.S. Glycogen storage disease: Clinical, biochemical, and molecular heterogeneity. Semin. Pediatr. Neurol. 2006, 13, 115–120.
- Arad, M.; Maron, B.J.; Gorham, J.M.; Johnson, W.H., Jr.; Saul, J.P.; Perez-Atayde, A.R.; Spirito, P.; Wright, G.B.; Kanter, R.J.; Seidman, C.E.; et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med. 2005, 352, 362–372.
- Wan, L.; Lee, C.C.; Hsu, C.M.; Hwu, W.L.; Yang, C.C.; Tsai, C.H.; Tsai, F.J. Identification of eight novel mutations of the acid α-glucosidase gene causing the infantile or juvenile form of glycogen storage disease type II. J. Neurol. 2008, 255, 831–838.
- Fernandez-Hojas, R.; Huie, M.L.; Navarro, C.; Dominguez, C.; Roig, M.; Lopez-Coronas, D.; Teijeira, S.; Anyane-Yeboa, K.; Hirschhorn, R. Identification of six novel mutations in the acid α-glucosidase gene in three Spanish patients with infantile onset glycogen storage disease type II (Pompe disease). Neuromuscul. Disord. NMD 2002, 12, 159–166.
- Cho, A.; Kim, S.J.; Lim, B.C.; Hwang, H.; Park, J.D.; Kim, G.B.; Jin, D.K.; Lee, J.; Ki, C.S.; Kim, K.J.; et al. Infantile Pompe disease: Clinical and genetic characteristics with an experience of enzyme replacement therapy. J. Child Neurol. 2012, 27, 319–324.
- Oitani, Y.; Ishiyama, A.; Kosuga, M.; Iwasawa, K.; Ogata, A.; Tanaka, F.; Takeshita, E.; Shimizu-Motohashi, Y.; Komaki, H.; Nishino, I.; et al. Interpretation of acid α-glucosidase activity in creatine kinase elevation: A case of Becker muscular dystrophy. Brain Dev. 2018, 40, 837–840.
- Laforet, P.; Nicolino, M.; Eymard, P.B.; Puech, J.P.; Caillaud, C.; Poenaru, L.; Fardeau, M. Juvenile and adult-onset acid maltase deficiency in France: Genotype-phenotype correlation. Neurology 2000, 55, 1122–1128.
- Muller-Felber, W.; Horvath, R.; Gempel, K.; Podskarbi, T.; Shin, Y.; Pongratz, D.; Walter, M.C.; Baethmann, M.; Schlotter-Weigel, B.; Lochmuller, H.; et al. Late onset Pompe disease: Clinical and neurophysiological spectrum of 38 patients including long-term follow-up in 18 patients. Neuromuscul. Disord. NMD 2007, 17, 698–706.
- Pittis, M.G.; Donnarumma, M.; Montalvo, A.L.; Dominissini, S.; Kroos, M.; Rosano, C.; Stroppiano, M.; Bianco, M.G.; Donati, M.A.; Parenti, G.; et al. Molecular and functional characterization of eight novel GAA mutations in Italian infants with Pompe disease. Hum. Mutat. 2008, 29, E27–E36.
- Nascimbeni, A.C.; Fanin, M.; Tasca, E.; Angelini, C. Molecular pathology and enzyme processing in various phenotypes of acid maltase deficiency. Neurology 2008, 70, 617–626.
- Boucek, D.; Jirikowic, J.; Taylor, M. Natural history of Danon disease. Genet. Med. Off. J. Am. Coll. Med. Genet. 2011, 13, 563–568.
- Momosaki, K.; Kido, J.; Yoshida, S.; Sugawara, K.; Miyamoto, T.; Inoue, T.; Okumiya, T.; Matsumoto, S.; Endo, F.; Hirose, S.; et al. Newborn screening for Pompe disease in Japan: Report and literature review of mutations in the GAA gene in Japanese and Asian patients. J. Hum. Genet. 2019, 64, 741–755.
- Dou, W.; Peng, C.; Zheng, J.; Gu, X.; Fu, L.; Martiniuk, F.; Sheng, H.Z. A novel missense mutation in the acid α-glucosidase gene causing the classic infantile form of Pompe disease. Clin. Chim. Acta Int. J. Clin. Chem. 2006, 374, 145–146.
- Oba-Shinjo, S.M.; da Silva, R.; Andrade, F.G.; Palmer, R.E.; Pomponio, R.J.; Ciociola, K.M.; Carvalho, M.S.; Gutierrez, P.S.; Porta, G.; Marrone, C.D.; et al. Pompe disease in a Brazilian series: Clinical and molecular analyses with identification of nine new mutations. J. Neurol. 2009, 256, 1881–1890.
- Loscher, W.N.; Huemer, M.; Stulnig, T.M.; Simschitz, P.; Iglseder, S.; Eggers, C.; Moser, H.; Moslinger, D.; Freilinger, M.; Lagler, F.; et al. Pompe disease in Austria: Clinical, genetic and epidemiological aspects. J. Neurol. 2018, 265, 159–164.
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910.
- Reddy, H.M.; Cho, K.A.; Lek, M.; Estrella, E.; Valkanas, E.; Jones, M.D.; Mitsuhashi, S.; Darras, B.T.; Amato, A.A.; Lidov, H.G.; et al. The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J. Hum. Genet. 2017, 62, 243–252.
- Mori, M.; Haskell, G.; Kazi, Z.; Zhu, X.; DeArmey, S.M.; Goldstein, J.L.; Bali, D.; Rehder, C.; Cirulli, E.T.; Kishnani, P.S. Sensitivity of whole exome sequencing in detecting infantile- and late-onset Pompe disease. Mol. Genet. Metab. 2017, 122, 189–197.
- Duzkale, H.; Shen, J.; McLaughlin, H.; Alfares, A.; Kelly, M.A.; Pugh, T.J.; Funke, B.H.; Rehm, H.L.; Lebo, M.S. A systematic approach to assessing the clinical significance of genetic variants. Clin. Genet. 2013, 84, 453–463.
- Hermans, M.M.; van Leenen, D.; Kroos, M.A.; Beesley, C.E.; Van Der Ploeg, A.T.; Sakuraba, H.; Wevers, R.; Kleijer, W.; Michelakakis, H.; Kirk, E.P.; et al. Twenty-two novel mutations in the lysosomal α-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II. Hum. Mutat. 2004, 23, 47–56.
- Kroos, M.; Hoogeveen-Westerveld, M.; Michelakakis, H.; Pomponio, R.; Van der Ploeg, A.; Halley, D.; Reuser, A.; GAA Database Consortium; Augoustides-Savvopoulou, P.; Ausems, M.; et al. Update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Hum. Mutat. 2012, 33, 1161–1165.
- Morales, A.; Anilkumar, A.C. Glycogen Storage Disease Type II. In StatPearls; Ineligible Companies: Treasure Island, FL, USA, 2023.
- Schoser, B. Pompe disease: What are we missing? Ann. Transl. Med. 2019, 7, 292.
- Hers, H.G. α-Glucosidase deficiency in generalized glycogenstorage disease (Pompe’s disease). Biochem. J. 1963, 86, 11–16.
- Ronzitti, G.; Collaud, F.; Laforet, P.; Mingozzi, F. Progress and challenges of gene therapy for Pompe disease. Ann. Transl. Med. 2019, 7, 287.
- Colella, P.; Mingozzi, F. Gene Therapy for Pompe Disease: The Time is now. Hum. Gene Ther. 2019, 30, 1245–1262.
- Musumeci, O.; Toscano, A. Diagnostic tools in late onset Pompe disease (LOPD). Ann. Transl. Med. 2019, 7, 286.
- Takikita, S.; Myerowitz, R.; Zaal, K.; Raben, N.; Plotz, P.H. Murine muscle cell models for Pompe disease and their use in studying therapeutic approaches. Mol. Genet. Metab. 2009, 96, 208–217.
- Raben, N.; Schreiner, C.; Baum, R.; Takikita, S.; Xu, S.; Xie, T.; Myerowitz, R.; Komatsu, M.; Van der Meulen, J.H.; Nagaraju, K.; et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder--murine Pompe disease. Autophagy 2010, 6, 1078–1089.
- Raben, N.; Wong, A.; Ralston, E.; Myerowitz, R. Autophagy and mitochondria in Pompe disease: Nothing is so new as what has long been forgotten. Am. J. Med. Genet. Part C Semin. Med. Genet. 2012, 160, 13–21.
- Rodriguez-Arribas, M.; Pedro, J.M.; Gomez-Sanchez, R.; Yakhine-Diop, S.M.; Martinez-Chacon, G.; Uribe-Carretero, E.; De Castro, D.C.; Casado-Naranjo, I.; Lopez de Munain, A.; Niso-Santano, M.; et al. Pompe Disease and Autophagy: Partners in Crime, or Cause and Consequence? Curr. Med. Chem. 2016, 23, 2275–2285.
- Endo, Y.; Furuta, A.; Nishino, I. Danon disease: A phenotypic expression of LAMP-2 deficiency. Acta Neuropathol. 2015, 129, 391–398.
- Stypmann, J.; Janssen, P.M.; Prestle, J.; Engelen, M.A.; Kogler, H.; Lullmann-Rauch, R.; Eckardt, L.; von Figura, K.; Landgrebe, J.; Mleczko, A.; et al. LAMP-2 deficient mice show depressed cardiac contractile function without significant changes in calcium handling. Basic Res. Cardiol. 2006, 101, 281–291.
- Chi, C.; Leonard, A.; Knight, W.E.; Beussman, K.M.; Zhao, Y.; Cao, Y.; Londono, P.; Aune, E.; Trembley, M.A.; Small, E.M.; et al. LAMP-2B regulates human cardiomyocyte function by mediating autophagosome-lysosome fusion. Proc. Natl. Acad. Sci. USA 2019, 116, 556–565.
- Hashem, S.I.; Murphy, A.N.; Divakaruni, A.S.; Klos, M.L.; Nelson, B.C.; Gault, E.C.; Rowland, T.J.; Perry, C.N.; Gu, Y.; Dalton, N.D.; et al. Impaired mitophagy facilitates mitochondrial damage in Danon disease. J. Mol. Cell. Cardiol. 2017, 108, 86–94.
- Olsen, A.S.B.; Faergeman, N.J. Sphingolipids: Membrane microdomains in brain development, function and neurological diseases. Open Biol. 2017, 7, 170069.
- Allende, M.L.; Zhu, H.; Kono, M.; Hoachlander-Hobby, L.E.; Huso, V.L.; Proia, R.L. Genetic defects in the sphingolipid degradation pathway and their effects on microglia in neurodegenerative disease. Cell. Signal. 2021, 78, 109879.
- Abed Rabbo, M.; Khodour, Y.; Kaguni, L.S.; Stiban, J. Sphingolipid lysosomal storage diseases: From bench to bedside. Lipids Health Dis. 2021, 20, 44.
- Tamboli, I.Y.; Hampel, H.; Tien, N.T.; Tolksdorf, K.; Breiden, B.; Mathews, P.M.; Saftig, P.; Sandhoff, K.; Walter, J. Sphingolipid storage affects autophagic metabolism of the amyloid precursor protein and promotes Aβ generation. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 1837–1849.
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71.
- De Castro-Oros, I.; Irun, P.; Cebolla, J.J.; Rodriguez-Sureda, V.; Mallen, M.; Pueyo, M.J.; Mozas, P.; Dominguez, C.; Pocovi, M.; Spanish, N.P.C.G. Assessment of plasma chitotriosidase activity, CCL18/PARC concentration and NP-C suspicion index in the diagnosis of Niemann-Pick disease type C: A prospective observational study. J. Transl. Med. 2017, 15, 43.
- Tammachote, R.; Tongkobpetch, S.; Srichomthong, C.; Phipatthanananti, K.; Pungkanon, S.; Wattanasirichaigoon, D.; Suphapeetiporn, K.; Shotelersuk, V. A common and two novel GBA mutations in Thai patients with Gaucher disease. J. Hum. Genet. 2013, 58, 594–599.
- Trilck, M.; Peter, F.; Zheng, C.; Frank, M.; Dobrenis, K.; Mascher, H.; Rolfs, A.; Frech, M.J. Diversity of glycosphingolipid GM2 and cholesterol accumulation in NPC1 patient-specific iPSC-derived neurons. Brain Res. 2017, 1657, 52–61.
- Oliveira, J.P.; Nowak, A.; Barbey, F.; Torres, M.; Nunes, J.P.; Teixeira, E.C.F.; Carvalho, F.; Sampaio, S.; Tavares, I.; Pereira, O.; et al. Fabry disease caused by the GLA p.Phe113Leu (p.F113L) variant: Natural history in males. Eur. J. Med. Genet. 2020, 63, 103703.
- Alharbi, F.J.; Baig, S.; Auray-Blais, C.; Boutin, M.; Ward, D.G.; Wheeldon, N.; Steed, R.; Dawson, C.; Hughes, D.; Geberhiwot, T. Globotriaosylsphingosine (Lyso-Gb(3)) as a biomarker for cardiac variant (N215S) Fabry disease. J. Inherit. Metab. Dis. 2018, 41, 239–247.
- Welford, R.W.D.; Muhlemann, A.; Garzotti, M.; Rickert, V.; Groenen, P.M.A.; Morand, O.; Uceyler, N.; Probst, M.R. Glucosylceramide synthase inhibition with lucerastat lowers globotriaosylceramide and lysosome staining in cultured fibroblasts from Fabry patients with different mutation types. Hum. Mol. Genet. 2018, 27, 3392–3403.
- Elmonem, M.A.; Mahmoud, I.G.; Mehaney, D.A.; Sharaf, S.A.; Hassan, S.A.; Orabi, A.; Salem, F.; Girgis, M.Y.; El-Badawy, A.; Abdelwahab, M.; et al. Lysosomal Storage Disorders in Egyptian Children. Indian J. Pediatr. 2016, 83, 805–813.
- Martinez-Archundia, M.; Hernandez Mojica, T.G.; Correa-Basurto, J.; Montano, S.; Camacho-Molina, A. Molecular dynamics simulations reveal structural differences among wild-type NPC1 protein and its mutant forms. J. Biomol. Struct. Dyn. 2020, 38, 3527–3532.
- Gomez-Grau, M.; Albaiges, J.; Casas, J.; Auladell, C.; Dierssen, M.; Vilageliu, L.; Grinberg, D. New murine Niemann-Pick type C models bearing a pseudoexon-generating mutation recapitulate the main neurobehavioural and molecular features of the disease. Sci. Rep. 2017, 7, 41931.
- Verot, L.; Chikh, K.; Freydiere, E.; Honore, R.; Vanier, M.T.; Millat, G. Niemann-Pick C disease: Functional characterization of three NPC2 mutations and clinical and molecular update on patients with NPC2. Clin. Genet. 2007, 71, 320–330.
- Patterson, M. Niemann-Pick Disease Type C. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Millat, G.; Chikh, K.; Naureckiene, S.; Sleat, D.E.; Fensom, A.H.; Higaki, K.; Elleder, M.; Lobel, P.; Vanier, M.T. Niemann-Pick disease type C: Spectrum of HE1 mutations and genotype/phenotype correlations in the NPC2 group. Am. J. Hum. Genet. 2001, 69, 1013–1021.
- Chikh, K.; Rodriguez, C.; Vey, S.; Vanier, M.T.; Millat, G. Niemann-Pick type C disease: Subcellular location and functional characterization of NPC2 proteins with naturally occurring missense mutations. Hum. Mutat. 2005, 26, 20–28.
- Kawazoe, T.; Yamamoto, T.; Narita, A.; Ohno, K.; Adachi, K.; Nanba, E.; Noguchi, A.; Takahashi, T.; Maekawa, M.; Eto, Y.; et al. Phenotypic variability of Niemann-Pick disease type C including a case with clinically pure schizophrenia: A case report. BMC Neurol. 2018, 18, 117.
- Barman, H.A.; Ikitimur, B.; Kilickiran Avci, B.; Durmaz, E.; Atici, A.; Aslan, S.; Ceylaner, S.; Karpuz, H. The Prevalence of Fabry Disease Among Turkish Patients with Non-Obstructive Hypertrophic Cardiomyopathy: Insights from a Screening Study. Balk. Med. J. 2019, 36, 354–358.
- Sheth, J.; Joseph, J.J.; Shah, K.; Muranjan, M.; Mistri, M.; Sheth, F. Pulmonary manifestations in Niemann-Pick type C disease with mutations in NPC2 gene: Case report and review of literature. BMC Med. Genet. 2017, 18, 5.
- Reunert, J.; Fobker, M.; Kannenberg, F.; Du Chesne, I.; Plate, M.; Wellhausen, J.; Rust, S.; Marquardt, T. Rapid Diagnosis of 83 Patients with Niemann Pick Type C Disease and Related Cholesterol Transport Disorders by Cholestantriol Screening. EBioMedicine 2016, 4, 170–175.
- Liu, R.; Zou, Y.; Hong, J.; Cao, M.; Cui, B.; Zhang, H.; Chen, M.; Shi, J.; Ning, T.; Zhao, S.; et al. Rare Loss-of-Function Variants in NPC1 Predispose to Human Obesity. Diabetes 2017, 66, 935–947.
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31.
- Cervera-Gaviria, M.; Alcantara-Ortigoza, M.A.; Gonzalez-Del Angel, A.; Moyers-Perez, P.; Legorreta-Ramirez, B.G.; Barrera-Carmona, N.; Cervera-Gaviria, J. An uncommon inheritance pattern in Niemann-Pick disease type C: Identification of probable paternal germline mosaicism in a Mexican family. BMC Neurol. 2016, 16, 147.
- Rohanizadegan, M.; Abdo, S.M.; O’Donnell-Luria, A.; Mihalek, I.; Chen, P.; Sanders, M.; Leeman, K.; Cho, M.; Hung, C.; Bodamer, O. Utility of rapid whole-exome sequencing in the diagnosis of Niemann-Pick disease type C presenting with fetal hydrops and acute liver failure. Cold Spring Harb. Mol. Case Stud. 2017, 3, a002147.
- Linari, S.; Castaman, G. Clinical manifestations and management of Gaucher disease. Clin. Cases Miner. Bone Metab. Off. J. Ital. Soc. Osteoporos. Miner. Metab. Skelet. Dis. 2015, 12, 157–164.
- Indellicato, R.; Trinchera, M. The Link between Gaucher Disease and Parkinson’s Disease Sheds Light on Old and Novel Disorders of Sphingolipid Metabolism. Int. J. Mol. Sci. 2019, 20, 3304.
- Charrow, J.; Andersson, H.C.; Kaplan, P.; Kolodny, E.H.; Mistry, P.; Pastores, G.; Rosenbloom, B.E.; Scott, C.R.; Wappner, R.S.; Weinreb, N.J.; et al. The Gaucher registry: Demographics and disease characteristics of 1698 patients with Gaucher disease. Arch. Intern. Med. 2000, 160, 2835–2843.
- Rosenbloom, B.E.; Weinreb, N.J. Gaucher disease: A comprehensive review. Crit. Rev. Oncog. 2013, 18, 163–175.
- Koprivica, V.; Stone, D.L.; Park, J.K.; Callahan, M.; Frisch, A.; Cohen, I.J.; Tayebi, N.; Sidransky, E. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am. J. Hum. Genet. 2000, 66, 1777–1786.
- Weiss, K.; Gonzalez, A.; Lopez, G.; Pedoeim, L.; Groden, C.; Sidransky, E. The clinical management of Type 2 Gaucher disease. Mol. Genet. Metab. 2015, 114, 110–122.
- Granek, Z.; Barczuk, J.; Siwecka, N.; Rozpedek-Kaminska, W.; Kucharska, E.; Majsterek, I. GBA1 Gene Mutations in α-Synucleinopathies-Molecular Mechanisms Underlying Pathology and Their Clinical Significance. Int. J. Mol. Sci. 2023, 24, 2044.
- Awad, O.; Sarkar, C.; Panicker, L.M.; Miller, D.; Zeng, X.; Sgambato, J.A.; Lipinski, M.M.; Feldman, R.A. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 2015, 24, 5775–5788.
- Sun, Y.; Liou, B.; Ran, H.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Kitatani, K.; Hannun, Y.A.; Witte, D.P.; Xu, Y.H.; et al. Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097.
- Brauer, A.U.; Kuhla, A.; Holzmann, C.; Wree, A.; Witt, M. Current Challenges in Understanding the Cellular and Molecular Mechanisms in Niemann-Pick Disease Type C1. Int. J. Mol. Sci. 2019, 20, 4392.
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140.
- Hendriksz, C.J.; Anheim, M.; Bauer, P.; Bonnot, O.; Chakrapani, A.; Corvol, J.C.; de Koning, T.J.; Degtyareva, A.; Dionisi-Vici, C.; Doss, S.; et al. The hidden Niemann-Pick type C patient: Clinical niches for a rare inherited metabolic disease. Curr. Med. Res. Opin. 2017, 33, 877–890.
- Sarkar, S.; Carroll, B.; Buganim, Y.; Maetzel, D.; Ng, A.H.; Cassady, J.P.; Cohen, M.A.; Chakraborty, S.; Wang, H.; Spooner, E.; et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013, 5, 1302–1315.
- Guo, H.; Zhao, M.; Qiu, X.; Deis, J.A.; Huang, H.; Tang, Q.Q.; Chen, X. Niemann-Pick type C2 deficiency impairs autophagy-lysosomal activity, mitochondrial function, and TLR signaling in adipocytes. J. Lipid Res. 2016, 57, 1644–1658.
- Yuasa, T.; Takenaka, T.; Higuchi, K.; Uchiyama, N.; Horizoe, Y.; Cyaen, H.; Mizukami, N.; Takasaki, K.; Kisanuki, A.; Miyata, M.; et al. Fabry disease. J. Echocardiogr. 2017, 15, 151–157.
- Turkmen, K.; Baloglu, I. Fabry disease: Where are we now? Int. Urol. Nephrol. 2020, 52, 2113–2122.
- Basta, M.; Pandya, A.M. Genetics, X-Linked Inheritance. In StatPearls; Ineligible Companies: Treasure Island, FL, USA, 2023.
- Felis, A.; Whitlow, M.; Kraus, A.; Warnock, D.G.; Wallace, E. Current and Investigational Therapeutics for Fabry Disease. Kidney Int. Rep. 2020, 5, 407–413.
- Breiden, B.; Sandhoff, K. Lysosomal Glycosphingolipid Storage Diseases. Annu. Rev. Biochem. 2019, 88, 461–485.
- du Moulin, M.; Muschol, N.P. D313Y is more than just a polymorphism in Fabry disease. Clin. Genet. 2018, 93, 1258.
- Capuano, I.; Garofalo, C.; Buonanno, P.; Pinelli, M.; Di Risi, T.; Feriozzi, S.; Riccio, E.; Pisani, A. Identifying Fabry patients in dialysis population: Prevalence of GLA mutations by renal clinic screening, 1995–2019. J. Nephrol. 2020, 33, 569–581.
- Chevrier, M.; Brakch, N.; Celine, L.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Annie Laquerriere, A.L.; Barbey, F.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599.
- Nelson, M.P.; Tse, T.E.; O’Quinn, D.B.; Percival, S.M.; Jaimes, E.A.; Warnock, D.G.; Shacka, J.J. Autophagy-lysosome pathway associated neuropathology and axonal degeneration in the brains of α-galactosidase A-deficient mice. Acta Neuropathol. Commun. 2014, 2, 20.
- Braun, F.; Blomberg, L.; Brodesser, S.; Liebau, M.C.; Schermer, B.; Benzing, T.; Kurschat, C.E. Enzyme Replacement Therapy Clears Gb3 Deposits from a Podocyte Cell Culture Model of Fabry Disease but Fails to Restore Altered Cellular Signaling. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2019, 52, 1139–1150.
- Ostergaard, J.R. Juvenile neuronal ceroid lipofuscinosis (Batten disease): Current insights. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 73–83.
- Mukherjee, A.B.; Appu, A.P.; Sadhukhan, T.; Casey, S.; Mondal, A.; Zhang, Z.; Bagh, M.B. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol. Neurodegener. 2019, 14, 4.
- Hersheson, J.; Burke, D.; Clayton, R.; Anderson, G.; Jacques, T.S.; Mills, P.; Wood, N.W.; Gissen, P.; Clayton, P.; Fearnley, J.; et al. Cathepsin D deficiency causes juvenile-onset ataxia and distinctive muscle pathology. Neurology 2014, 83, 1873–1875.
- Steinfeld, R.; Reinhardt, K.; Schreiber, K.; Hillebrand, M.; Kraetzner, R.; Bruck, W.; Saftig, P.; Gartner, J. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am. J. Hum. Genet. 2006, 78, 988–998.
- Siintola, E.; Partanen, S.; Stromme, P.; Haapanen, A.; Haltia, M.; Maehlen, J.; Lehesjoki, A.E.; Tyynela, J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain J. Neurol. 2006, 129, 1438–1445.
- Smith, K.R.; Dahl, H.H.; Canafoglia, L.; Andermann, E.; Damiano, J.; Morbin, M.; Bruni, A.C.; Giaccone, G.; Cossette, P.; Saftig, P.; et al. Cathepsin F mutations cause Type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2013, 22, 1417–1423.
- Wisniewski, K.E.; Zhong, N.; Kaczmarski, W.; Kaczmarski, A.; Kida, E.; Brown, W.T.; Schwarz, K.O.; Lazzarini, A.M.; Rubin, A.J.; Stenroos, E.S.; et al. Compound heterozygous genotype is associated with protracted juvenile neuronal ceroid lipofuscinosis. Ann. Neurol. 1998, 43, 106–110.
- Topcu, M.; Tan, H.; Yalnizoglu, D.; Usubutun, A.; Saatci, I.; Aynaci, M.; Anlar, B.; Topaloglu, H.; Turanli, G.; Kose, G.; et al. Evaluation of 36 patients from Turkey with neuronal ceroid lipofuscinosis: Clinical, neurophysiological, neuroradiological and histopathologic studies. Turk. J. Pediatr. 2004, 46, 1–10.
- Wheeler, R.B.; Sharp, J.D.; Schultz, R.A.; Joslin, J.M.; Williams, R.E.; Mole, S.E. The gene mutated in variant late-infantile neuronal ceroid lipofuscinosis (CLN6) and in nclf mutant mice encodes a novel predicted transmembrane protein. Am. J. Hum. Genet. 2002, 70, 537–542.
- Gao, H.; Boustany, R.M.; Espinola, J.A.; Cotman, S.L.; Srinidhi, L.; Antonellis, K.A.; Gillis, T.; Qin, X.; Liu, S.; Donahue, L.R.; et al. Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am. J. Hum. Genet. 2002, 70, 324–335.
- Kousi, M.; Siintola, E.; Dvorakova, L.; Vlaskova, H.; Turnbull, J.; Topcu, M.; Yuksel, D.; Gokben, S.; Minassian, B.A.; Elleder, M.; et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain J. Neurol. 2009, 132, 810–819.
- Aiello, C.; Terracciano, A.; Simonati, A.; Discepoli, G.; Cannelli, N.; Claps, D.; Crow, Y.J.; Bianchi, M.; Kitzmuller, C.; Longo, D.; et al. Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum. Mutat. 2009, 30, E530–E540.
- Aldahmesh, M.A.; Al-Hassnan, Z.N.; Aldosari, M.; Alkuraya, F.S. Neuronal ceroid lipofuscinosis caused by MFSD8 mutations: A common theme emerging. Neurogenetics 2009, 10, 307–311.
- Mole, S.E.; Williams, R.E. Neuronal Ceroid-Lipofuscinoses—Retired Chapter, for Historical Reference Only. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Cortese, A.; Tucci, A.; Piccolo, G.; Galimberti, C.A.; Fratta, P.; Marchioni, E.; Grampa, G.; Cereda, C.; Grieco, G.; Ricca, I.; et al. Novel CLN3 mutation causing autophagic vacuolar myopathy. Neurology 2014, 82, 2072–2076.
- Cannelli, N.; Cassandrini, D.; Bertini, E.; Striano, P.; Fusco, L.; Gaggero, R.; Specchio, N.; Biancheri, R.; Vigevano, F.; Bruno, C.; et al. Novel mutations in CLN8 in Italian variant late infantile neuronal ceroid lipofuscinosis: Another genetic hit in the Mediterranean. Neurogenetics 2006, 7, 111–117.
- Teixeira, C.A.; Espinola, J.; Huo, L.; Kohlschutter, J.; Persaud Sawin, D.A.; Minassian, B.; Bessa, C.J.; Guimaraes, A.; Stephan, D.A.; Sa Miranda, M.C.; et al. Novel mutations in the CLN6 gene causing a variant late infantile neuronal ceroid lipofuscinosis. Hum. Mutat. 2003, 21, 502–508.
- Siintola, E.; Topcu, M.; Aula, N.; Lohi, H.; Minassian, B.A.; Paterson, A.D.; Liu, X.Q.; Wilson, C.; Lahtinen, U.; Anttonen, A.K.; et al. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am. J. Hum. Genet. 2007, 81, 136–146.
- Sarpong, A.; Schottmann, G.; Ruther, K.; Stoltenburg, G.; Kohlschutter, A.; Hubner, C.; Schuelke, M. Protracted course of juvenile ceroid lipofuscinosis associated with a novel CLN3 mutation (p.Y199X). Clin. Genet. 2009, 76, 38–45.
- Di Fabio, R.; Moro, F.; Pestillo, L.; Meschini, M.C.; Pezzini, F.; Doccini, S.; Casali, C.; Pierelli, F.; Simonati, A.; Santorelli, F.M. Pseudo-dominant inheritance of a novel CTSF mutation associated with type B Kufs disease. Neurology 2014, 83, 1769–1770.
- Munroe, P.B.; Mitchison, H.M.; O’Rawe, A.M.; Anderson, J.W.; Boustany, R.M.; Lerner, T.J.; Taschner, P.E.; de Vos, N.; Breuning, M.H.; Gardiner, R.M.; et al. Spectrum of mutations in the Batten disease gene, CLN3. Am. J. Hum. Genet. 1997, 61, 310–316.
- Siintola, E.; Topcu, M.; Kohlschutter, A.; Salonen, T.; Joensuu, T.; Anttonen, A.K.; Lehesjoki, A.E. Two novel CLN6 mutations in variant late-infantile neuronal ceroid lipofuscinosis patients of Turkish origin. Clin. Genet. 2005, 68, 167–173.
- Cherot, E.; Keren, B.; Dubourg, C.; Carre, W.; Fradin, M.; Lavillaureix, A.; Afenjar, A.; Burglen, L.; Whalen, S.; Charles, P.; et al. Using medical exome sequencing to identify the causes of neurodevelopmental disorders: Experience of 2 clinical units and 216 patients. Clin. Genet. 2018, 93, 567–576.
- Ranta, S.; Topcu, M.; Tegelberg, S.; Tan, H.; Ustubutun, A.; Saatci, I.; Dufke, A.; Enders, H.; Pohl, K.; Alembik, Y.; et al. Variant late infantile neuronal ceroid lipofuscinosis in a subset of Turkish patients is allelic to Northern epilepsy. Hum. Mutat. 2004, 23, 300–305.
- Kim, W.D.; Wilson-Smillie, M.; Thanabalasingam, A.; Lefrancois, S.; Cotman, S.L.; Huber, R.J. Autophagy in the Neuronal Ceroid Lipofuscinoses (Batten Disease). Front. Cell Dev. Biol. 2022, 10, 812728.
- Stepien, K.M.; Roncaroli, F.; Turton, N.; Hendriksz, C.J.; Roberts, M.; Heaton, R.A.; Hargreaves, I. Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. J. Clin. Med. 2020, 9, 2596.
- Anderson, G.W.; Goebel, H.H.; Simonati, A. Human pathology in NCL. Biochim. Biophys. Acta 2013, 1832, 1807–1826.
- Chen, J.; Soni, R.K.; Xu, Y.; Simoes, S.; Liang, F.X.; DeFreitas, L.; Hwang, R., Jr.; Montesinos, J.; Lee, J.H.; Area-Gomez, E.; et al. Juvenile CLN3 disease is a lysosomal cholesterol storage disorder: Similarities with Niemann-Pick type C disease. EBioMedicine 2023, 92, 104628.
- Mirza, M.; Vainshtein, A.; DiRonza, A.; Chandrachud, U.; Haslett, L.J.; Palmieri, M.; Storch, S.; Groh, J.; Dobzinski, N.; Napolitano, G.; et al. The CLN3 gene and protein: What we know. Mol. Genet. Genom. Med. 2019, 7, e859.
- Cotman, S.L.; Vrbanac, V.; Lebel, L.A.; Lee, R.L.; Johnson, K.A.; Donahue, L.R.; Teed, A.M.; Antonellis, K.; Bronson, R.T.; Lerner, T.J.; et al. Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum. Mol. Genet. 2002, 11, 2709–2721.
- Cao, Y.; Espinola, J.A.; Fossale, E.; Massey, A.C.; Cuervo, A.M.; MacDonald, M.E.; Cotman, S.L. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J. Biol. Chem. 2006, 281, 20483–20493.
- Zheng, W.; Chen, Q.; Wang, C.; Yao, D.; Zhu, L.; Pan, Y.; Zhang, J.; Bai, Y.; Shao, C. Inhibition of Cathepsin D (CTSD) enhances radiosensitivity of glioblastoma cells by attenuating autophagy. Mol. Carcinog. 2020, 59, 651–660.
- Michalski, J.C.; Klein, A. Glycoprotein lysosomal storage disorders: α- and β-mannosidosis, fucosidosis and α-N-acetylgalactosaminidase deficiency. Biochim. Biophys. Acta 1999, 1455, 69–84.
- Malm, D.; Stensland, H.M.F.R.; Nilssen, Ø. Glycoproteinoses. In Lysosomal Storage Disorders; Frontiers: Lausanne, Switzerland, 2022; pp. 203–210.
- Cantz, M.; Ulrich-Bott, B. Disorders of glycoprotein degradation. J. Inherit. Metab. Dis. 1990, 13, 523–537.
- Bedilu, R.; Nummy, K.A.; Cooper, A.; Wevers, R.; Smeitink, J.; Kleijer, W.J.; Friderici, K.H. Variable clinical presentation of lysosomal β-mannosidosis in patients with null mutations. Mol. Genet. Metab. 2002, 77, 282–290.
- Berg, T.; Riise, H.M.; Hansen, G.M.; Malm, D.; Tranebjaerg, L.; Tollersrud, O.K.; Nilssen, O. Spectrum of mutations in α-mannosidosis. Am. J. Hum. Genet. 1999, 64, 77–88.
- Borgwardt, L.; Stensland, H.M.; Olsen, K.J.; Wibrand, F.; Klenow, H.B.; Beck, M.; Amraoui, Y.; Arash, L.; Fogh, J.; Nilssen, O.; et al. α-mannosidosis: Correlation between phenotype, genotype and mutant MAN2B1 subcellular localisation. Orphanet J. Rare Dis. 2015, 10, 70.
- Gotoda, Y.; Wakamatsu, N.; Kawai, H.; Nishida, Y.; Matsumoto, T. Missense and nonsense mutations in the lysosomal α-mannosidase gene (MANB) in severe and mild forms of α-mannosidosis. Am. J. Hum. Genet. 1998, 63, 1015–1024.
- Hossain, M.A.; Higaki, K.; Shinpo, M.; Nanba, E.; Suzuki, Y.; Ozono, K.; Sakai, N. Chemical chaperone treatment for galactosialidosis: Effect of NOEV on β-galactosidase activities in fibroblasts. Brain Dev. 2016, 38, 175–180.
- Khan, J.M.; Ranganathan, S. A multi-species comparative structural bioinformatics analysis of inherited mutations in α-D-mannosidase reveals strong genotype-phenotype correlation. BMC Genom. 2009, 10 (Suppl. 3), S33.
- Prada, C.E.; Gonzaga-Jauregui, C.; Tannenbaum, R.; Penney, S.; Lupski, J.R.; Hopkin, R.J.; Sutton, V.R. Clinical utility of whole-exome sequencing in rare diseases: Galactosialidosis. Eur. J. Med. Genet. 2014, 57, 339–344.
- Riise Stensland, H.M.; Klenow, H.B.; Van Nguyen, L.; Hansen, G.M.; Malm, D.; Nilssen, O. Identification of 83 novel α-mannosidosis-associated sequence variants: Functional analysis of MAN2B1 missense mutations. Hum. Mutat. 2012, 33, 511–520.
- Riise Stensland, H.M.F.; Persichetti, E.; Sorriso, C.; Hansen, G.M.; Bibi, L.; Paciotti, S.; Balducci, C.; Beccari, T. Identification of two novel β-mannosidosis-associated sequence variants: Biochemical analysis of β-mannosidase (MANBA) missense mutations. Mol. Genet. Metab. 2008, 94, 476–480.
- Seo, H.C.; Willems, P.J.; O’Brien, J.S. Six additional mutations in fucosidosis: Three nonsense mutations and three frameshift mutations. Hum. Mol. Genet. 1993, 2, 1205–1208.
- Shimmoto, M.; Fukuhara, Y.; Itoh, K.; Oshima, A.; Sakuraba, H.; Suzuki, Y. Protective protein gene mutations in galactosialidosis. J. Clin. Investig. 1993, 91, 2393–2398.
- Takiguchi, K.; Itoh, K.; Shimmoto, M.; Ozand, P.T.; Doi, H.; Sakuraba, H. Structural and functional study of K453E mutant protective protein/cathepsin A causing the late infantile form of galactosialidosis. J. Hum. Genet. 2000, 45, 200–206.
- Tiberio, G.; Filocamo, M.; Gatti, R.; Durand, P. Mutations in fucosidosis gene: A review. Acta Genet. Med. Gemellol. 1995, 44, 223–232.
- Willems, P.J.; Seo, H.C.; Coucke, P.; Tonlorenzi, R.; O’Brien, J.S. Spectrum of mutations in fucosidosis. Eur. J. Hum. Genet. 1999, 7, 60–67.
- Zhou, X.Y.; van der Spoel, A.; Rottier, R.; Hale, G.; Willemsen, R.; Berry, G.T.; Strisciuglio, P.; Morrone, A.; Zammarchi, E.; Andria, G.; et al. Molecular and biochemical analysis of protective protein/cathepsin A mutations: Correlation with clinical severity in galactosialidosis. Hum. Mol. Genet. 1996, 5, 1977–1987.
- Adam, J.; Malone, R.; Lloyd, S.; Lee, J.; Hendriksz, C.J.; Ramaswami, U. Disease progression of α-mannosidosis and impact on patients and carers—A UK natural history survey. Mol. Genet. Metab. Rep. 2019, 20, 100480.
- Bertolini, A.; Rigoldi, M.; Cianflone, A.; Mariani, R.; Piperno, A.; Canonico, F.; Cefalo, G.; Carubbi, F.; Simonati, A.; Urban, M.L.; et al. Long-term outcome of a cohort of Italian patients affected with α-Mannosidosis. Clin. Dysmorphol. 2024, 33, 1–8.
- Verrecchia, E.; Sicignano, L.L.; Massaro, M.G.; Rocco, R.; Silvestri, G.; Rossi, S.; Manna, R. Caregivers’ and Physicians’ Perspectives on α-Mannosidosis: A Report from Italy. Adv. Ther. 2021, 38, 1–10.
- Malm, D.; Nilssen, O. α-mannosidosis. Orphanet J. Rare Dis. 2008, 3, 21.
- Ceccarini, M.R.; Codini, M.; Conte, C.; Patria, F.; Cataldi, S.; Bertelli, M.; Albi, E.; Beccari, T. α-Mannosidosis: Therapeutic Strategies. Int. J. Mol. Sci. 2018, 19, 1500.
- Borgwardt, L.G.; Ceravolo, F.; Zardi, G.; Ballabeni, A.; Lund, A.M. Relationship between MAN2B1 genotype/subcellular localization subgroups, antidrug antibody detection, and long-term velmanase alfa treatment outcomes in patients with α-mannosidosis. JIMD Rep. 2023, 64, 187–198.
- Labauge, P.; Renard, D.; Castelnovo, G.; Sabourdy, F.; de Champfleur, N.; Levade, T. β-mannosidosis: A new cause of spinocerebellar ataxia. Clin. Neurol. Neurosurg. 2009, 111, 109–110.
- Cooper, A.; Hatton, C.E.; Thornley, M.; Sardharwalla, I.B. α- and β-mannosidoses. J. Inherit. Metab. Dis. 1990, 13, 538–548.
- Gowda, V.K.; Nagarajan, B.; Suryanarayana, S.G.; Srinivasan, V.M. Familial Global Developmental Delay Secondary to β-Mannosidosis. J. Pediatr. Neurosci. 2021, 16, 149–152.
- He, R.; Liu, J.; Xu, H.; Tang, X.; Liu, H.; Zhao, S. β-mannosidosis presenting predominantly with recurrent pulmonary infections, hemorrhage, and cystic lesions. Pediatr. Pulmonol. 2023, 58, 1272–1274.
- Blomqvist, M.; Smeland, M.F.; Lindgren, J.; Sikora, P.; Riise Stensland, H.M.F.; Asin-Cayuela, J. β-Mannosidosis caused by a novel homozygous intragenic inverted duplication in MANBA. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003954.
- Gytz, H.; Liang, J.; Liang, Y.; Gorelik, A.; Illes, K.; Nagar, B. The structure of mammalian β-mannosidase provides insight into β-mannosidosis and nystagmus. FEBS J. 2019, 286, 1319–1331.
More
Information
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
998
Revisions:
2 times
(View History)
Update Date:
10 Jan 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No