1. Introduction
Lysosomal storage disorders (LSDs) arise from abnormal lysosomal function, leading to the accumulation of undegraded metabolites. The specific composition of these materials accumulated in lysosomes varies significantly among LSDs. Given the relevance of the lysosomal pathway in cellular homeostasis, its malfunction leads to the dysregulation of several cellular processes linked to this organelle such as lipid homeostasis, cell viability, exocytosis, membrane repair, and autophagy, among others [
40]. Lysosomes are the final scenario of autophagic degradation, and the inability to clear autophagosomes results in the accumulation of undesired cargo that can further hinder cell viability. Thus, it is not surprising that defective autophagy has already been described in different LSDs [
41], contributing to the development of the disease.
2. Mucopolysaccharidoses
Mucopolysaccharidoses (MPS) are a group of inherited diseases caused by mutations in genes encoding several lysosomal enzymes (including ARSB, GALNS, GLB1, GNS, GUSB, HGSNAT, HYAL1, IDS, IDUA, NAGLU, and SGSH), which are involved in the degradation of glycosaminoglycans (GAGs). However, it was described that additional alterations in other pathways (including autophagy) play a significant role in the pathogenesis of these diseases [
42]. In this regard, GAGs are linear polysaccharides composed of repeating disaccharide units, which are highly sulphated. This heterogeneous group includes molecules like hyaluronic acid, heparan sulphate, dermatan sulphate, or keratan sulphate, and they are present in all human tissues. Therefore, accumulation of GAGs affects a wide range of organs and systems, and the CNS is the most commonly impacted. MPS patients exhibit a range of symptoms that differ in severity but share certain characteristic traits. These features involve facial characteristics, skeletal abnormalities, and multiorganic affectation such as heart issues, respiratory problems, and the enlargement of the liver and spleen. Thirteen different types and subtypes of MPSs have been described: MPS I (which includes three subtypes: Hurler, Hurler–Scheie, and Scheie syndromes), MPS II (also known as Hunter syndrome), MPS III (subtypes IIIA, IIIB, IIIC, and IIID), MPS IV (subtypes IVA and IVB), MPS VI, MPS VII, and MPS IX [
43]. Mutations related to NCLs are listed in
Supplementary Table S1 [
44,
45,
46,
47,
48,
49,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77,
78,
79,
80,
81,
82,
83,
84,
85,
86,
87,
88,
89,
90,
91,
92,
93,
94,
95,
96,
97,
98,
99,
100,
101,
102,
103,
104,
105,
106,
107,
108,
109,
110,
111,
112,
113,
114,
115,
116,
117,
118,
119,
120,
121,
122,
123,
124,
125,
126,
127,
128,
129,
130,
131].
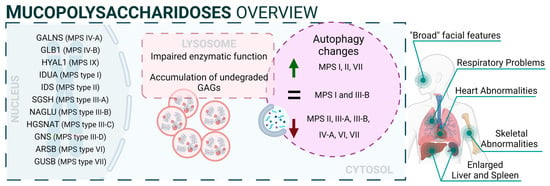
From a mechanistic point of view, autophagy levels were recently addressed in different models of MPS, resulting in conflicting conclusions [
132]. For example, impaired autophagy was described or suggested in MPS II [
133], MPS III-A [
134], MPS III-B [
135], MPS IV-A [
136], MPS VI [
137], and MPS VII [
138]. However, other reports also showed unaltered autophagy in MPS I and III-B [
139,
140] or even enhanced autophagic response in MPS I, II, and VII [
141,
142]. Though we cannot rule out the possibility of differential autophagy regulation in MPS subtypes, a possible explanation for this controversy could be the lack of complete, comprehensive autophagy flux analyses, which hinders the precise characterization of the autophagic pathway in these diseases, as has already been discussed in MPS III-C [
143]. Interestingly, Kondo and collaborators described a new MPS-like disorder caused by a mutation in VPS33A [
144], a gene that encodes for a protein that mediates autophagosome–lysosome fusion. However, this mutation (c.1492C > T; p.Arg498Trp) does not compromise the autophagy-related role of VPS33A, unveiling a new function of this protein (
Figure 2: mucopolysaccharidoses).
Figure 2. Mucopolysaccharidoses overview.
3. Autophagy in Glycogenoses
The glycogenoses are LSDs characterized by severe autophagy defects specifically affecting skeletal and cardiac muscles. In this regard, these diseases are commonly referred to as autophagic vacuolar myopathies (AVMs), although neuropathological manifestations are also observed in some particular cases [
145]. Conditions leading to AVM are associated with genetic mutations affecting genes responsible for glycogen hydrolysis, lysosome acidification, and the maturation and fusion of autophagosomes with lysosomes [
146]. Mutations related to glycogenoses are listed in Supplementary Table S2 [
147,
148,
149,
150,
151,
152,
153,
154,
155,
156,
157,
158,
159,
160,
161,
162,
163,
164,
165,
166,
167,
168,
169,
170,
171,
172,
173,
174,
175,
176].
3.1. Pompe Disease
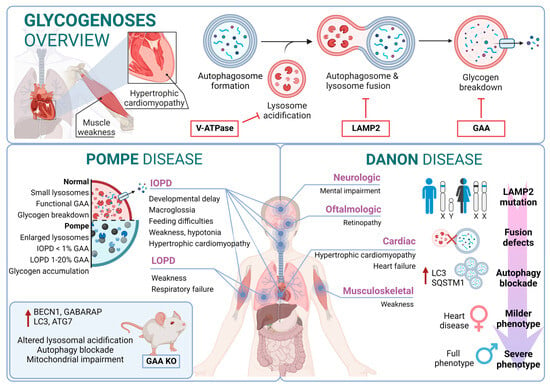
Pompe disease is a rare disorder caused by autosomal recessive mutations in the GAA gene that result in the accumulation of glycogen within lysosomes [
177,
178]. It was the first recognized LSD [
5] and is also known as glycogen storage disease type II (GSDII). Different studies established an association between specific GAA single-nucleotide polymorphisms (SNPs) and Pompe disease, including rs140826989, rs121907938, rs121907945, rs121907936, rs147804176, rs1555600061, rs1800312, and rs200856561, among others [
155,
170,
173]. Clinically, Pompe disease includes two main groups: infantile-onset Pompe disease (IOPD) and late-onset Pompe disease (LOPD) [
179]. IOPD patients typically exhibit severe symptoms within the first few months of life, including weakness, developmental delay, feeding difficulties, hypotonia, macroglossia, and hypertrophic cardiomyopathy. These patients experience multisystem glycogen accumulation with less than 1% of normal GAA enzyme function. Without treatment, their life expectancy is typically less than two years [
180].
On the other hand, LOPD patients may present symptoms anywhere from childhood to adulthood, with worse prognosis when symptoms manifest at an earlier age. These patients generally retain 1–20% of normal GAA function. However, abnormal glycogen build-up in the respiratory system can lead to respiratory failure, necessitating mechanical ventilation in a significant percentage of patients [
181]. From a molecular perspective, the glycogen accumulation interferes with cellular processes such as metabolism or autophagy. In this context, the accumulation of autophagy substrates and autophagosomes was demonstrated by using knockout mice models of GAA [
182,
183]. In fact, components of the autophagy system, including BECN1, GABARAP, LC3, and ATG7, are increased; however, this excess is associated with functional autophagy deficiency, as the machinery is unable to degrade them. Furthermore, in primary cells derived from KO mice, lysosomal acidification is inefficient, leading to a blockade of autophagy and an impairment of mitochondrial function, which are associated with this lysosomal disorder [
184,
185].
3.2. Danon Disease
Danon disease was the first LSD in which an association with autophagy involvement was reported. Danon disease it is characterized by a deficiency in LAMP2 (lysosomal-associated membrane protein 2). The disease is inherited as an X-linked trait and is exceptionally rare [
186]. From a phenotypic point of view, Danon disease is characterized by severe hypertrophic cardiomyopathy, heart failure, muscle weakness, retinopathy, and different degrees of mental retardation specifically in male patients. However, in female patients, it is described as a “milder phenotype” that is mostly limited to cardiac abnormalities.
According to its relation with the autophagic process, the accumulation of large LC3-positive membrane-bound structures is a very common feature in several tissues, especially in muscular tissues. In this context, an increase in lipofuscin accumulation and myofibrillar disruption are present in cardiac muscle too.
From a molecular point of view, most Danon patients carry mutations that result in LAMP2 (lysosomal-associated membrane protein 2) loss of function. In fact, Danon disease is also known as “glycogen storage disease due to LAMP2 deficiency”. These alterations cause an important failure in autophagosomal–lysosomal fusion accompanied by an excessive accumulation of autophagosomes and partial modifications in a subset of lysosomal enzymes and p62 aggregates. In this regard, by using muscle biopsies from patients, a correlation was reported between a blockage in autophagy flux and disease severity. In addition, mice deficient in Lamp2 mimic Danon disease in humans, showing an accumulation of autophagosomes, which severely affects cardiac contractile function [
187].
Although the most Danon-affected people present a deficiency of all three LAMP-2 isoforms, the pathogenesis and clinical manifestations are attributed to the specific deficiency of LAMP-2B, the expression of which is abundant in the heart, muscle, and brain. Indeed, only a defect in LAMP-2B is sufficient to cause most of the disease manifestations [
188]. Furthermore, these patients present defects also in mitochondrial clearance (mitophagy), which shows that it is not only bulk autophagy that is altered in this type of disease [
189] (
Figure 3).
Figure 3. Glycogenoses overview. Pompe disease and Danon disease.
4. Autophagic Process in Sphingolipidoses
Sphingolipids represent a major category of lipids in the nervous system and play an important role in neural development and functionality [
190]. Their metabolism is tightly regulated through a multistep degradation process that relies on several lysosomal hydrolases [
191]. In this regard, sphingolipidoses represent a broad group of inherited disorders related to sphingolipid metabolism that frequently affect the nervous system. Specifically, sphingolipidoses are a subset of LSDs characterized by the accumulation of partially or completely undegraded sphingolipids. These alterations are observed mainly in the pediatric population, manifesting in neurodegeneration that leads to psychomotor retardation and myoclonus due to widespread and progressive damage to neurons. In some cases, these conditions may cause weakness and spasticity due to the involvement of white-matter tracts. The genetic changes underlying these disorders are diverse and can lead to the accumulation of substances such as sphingomyelin, glycolipids, glucocerebrosides, gangliosides, unesterified cholesterol, and sulfatide compounds, among others [
192]. For these reasons, it is not surprising that several disturbances in autophagy have been reported in sphingolipidoses. For example, the addition of glycosphingolipids to cells, even by a simple method such as supplementation in a culture medium, triggers autophagy, prevents the clearance of autophagosomes, and leads to their accumulation [
193]. Diseases in this category include Niemann–Pick, Gaucher, and Fabry diseases as well as mucolipidoses, and GM1/2 gangliosidoses such as Tay–Sachs and Sandhoff diseases [
194]. Mutations related to sphingolipidoses are listed in Supplementary Table S3 [
195,
196,
197,
198,
199,
200,
201,
202,
203,
204,
205,
206,
207,
208,
209,
210,
211,
212,
213,
214,
215].
4.1. Gaucher Disease
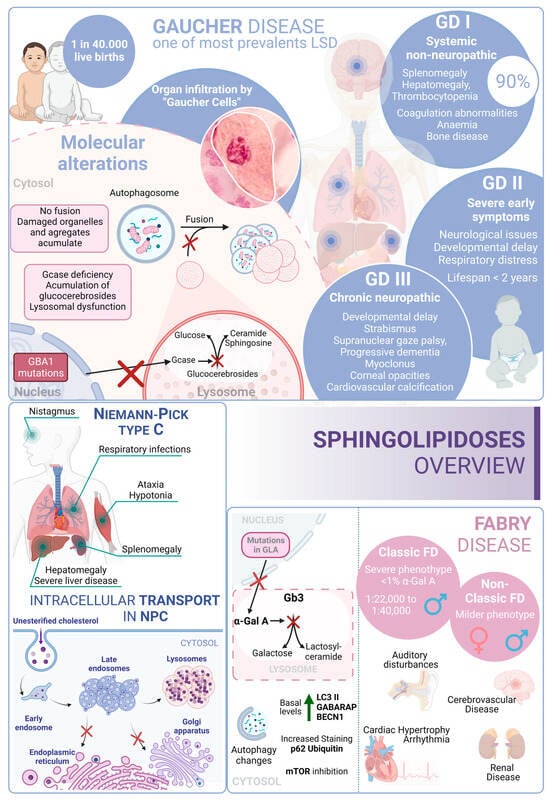
Gaucher disease (GD) is one of the most common lysosomal storage disorders and occurs in up to 1 in 40,000 live births in the general population. GD disease includes three clinical phenotypes: GD types I, II, and III [
216]. GD type I is considered a systemic disorder, with no neurological involvement. This type represents more than 90% of GD cases; the common symptoms are splenomegaly, hepatomegaly, thrombocytopenia, coagulation abnormalities, anemia, bone disease, and bone marrow infiltration by storage cells. The age of onset is highly variable and can start at any time from childhood to 70 years old. However, most patients are diagnosed before the age of 20 [
217].
Several studies pointed to a genetic association between the p.N370S allele, both homozygous and heterozygous states, and the development of GD type I [
218]. In contrast, GD type II often manifests with severe symptoms within the first six months of life. Patients with this form of the disease experience neurological manifestations, brainstem involvement, developmental delays, cachexia, respiratory distress, pneumonia, skin issues, and other symptoms. The prognosis for individuals with GD type II is very poor, as infants rapidly deteriorate, and unfortunately, most do not survive beyond 2 years of age [
219]. GD type III (commonly known as chronic neuropathic form) has a more insidious course. The manifestations are developmental delay; strabismus and supranuclear gaze palsy; progressive dementia; myoclonus; corneal opacities; and cardiovascular calcification. From a genetic point of view, it is known that individuals who are homozygous for the p.L444P allele probably develop type II or III [
220,
221]. In this regard, glucosylceramide and glucosylsphingosine are the primary glycosphingolipids that accumulate in this disease, primarily in macrophages known as “Gaucher cells”, which are found in the liver, spleen, lungs, and central nervous system. From a molecular point of view, these accumulations result from mutations in the GBA1 gene, which encodes glucocerebrosidase [
222].
In GD, there is an impairment of the autophagic process. In fact, it was described as an accumulation of autophagosomes in different in vitro and in vivo models. For example, using induced pluripotent stem cells (iPSCs) derived from Gaucher patient cells that were reprogrammed into neurons, the researchers observed an increase in the number of autophagosomes and the amount of autophagic markers such as LC3-II and p62, which appear exclusively in neuropathic cells. Specifically, affected neuropathic cells display blocked autophagic flux with reduced autophagosomal clearance and decreased levels of LAMP1 and TFEB. This indicates a lysosomal dysfunction and is a probable cause of neurodegeneration in this pathology. The overexpression of TFEB in combination with recombinant glucocerebrosidase treatment ameliorates these alterations [
223].
Furthermore, a mouse model that phenocopies GD mediated by mutations in both glucocerebrosidase (V394L) and C-saposin deficiencies shows accumulation of p62/SQSTM1 in neurons and astrocytes along with sequestration of undigested materials within axonal vesicles. These observations indicate that autolysosomal cargo degradation is impaired in cells affected by GD [
224].
In summary, autophagy dysfunction is present in various model systems of GD, but the underlying mechanisms are still unclear.
4.2. Niemann–Pick Type C Disease (NPC)
Niemann–Pick type C (NPC) disease is a genetic autosomal recessive lysosomal storage disorder caused by mutations in either NPC1 (95% of cases) or NPC2 (5% of cases). These genes encode proteins involved in the intracellular trafficking of lipids and cholesterol [
225]. Mutations in these genes result in the accumulation of unesterified cholesterol in the liver, spleen, and brain, which, in turn, disrupts lipid transport. In fact, these alterations cause a disruption that leads to the loss of Purkinje cells in the cerebellum and degeneration of other components of the central nervous system [
226].
From a clinical perspective, NPC is typically a disease with juvenile or later onset, and the rate of progression inversely correlates with the age of onset. Common symptoms of NPC include ataxia, splenomegaly, hepatomegaly, hypotonia, severe liver disease, respiratory infections, and abnormal eye movements [
227].
In this context, there is an alteration of the autophagic mechanism in NPC, as an accumulation of autophagosomes in skin fibroblasts from NPC patients is described. In the molecular landscape of this disease, this accumulation is partially due to the function of BECN1 and LC3B. In wild-type fibroblasts, their levels increase when exposed to U18666A, a small molecule used to induce NPC-like lipid trafficking defects. Moreover, NPC exhibits a blocked autophagic flux due to impaired autophagosome maturation [
228] and specific defects in mitophagy [
229].
Therefore, autophagy is significantly disrupted in NPC. This disruption interferes in the maintenance of cellular and tissue homeostasis, contributing to the pathological changes observed in NPC patients. The accumulation of autophagosomes, their impaired maturation, and the defective mitochondrial function all contribute to the disease’s progression, affecting cellular and tissue health.
4.3. Fabry Disease
Fabry disease (FD) is an X-linked LSD characterized by mutations in the GLA gene. This gene encodes the lysosomal enzyme α-galactosidase A (α-Gal A). Deficiency of α-Gal A causes an accumulation of globotriaosylceramide (Gb3) in organs, including the heart, kidneys, brain, and eyes, among others [
230]. FD is among the more frequent LSDs after GD [
231]. Due to its X-linked inheritance pattern, prevalence in males is higher than in females. In males, there are two subtypes: the classic presentation with a severe phenotype and the nonclassic with a less severe phenotype [
232]. The classical phenotype of FD has an incidence of 1:22,000 to 1:40,000 in males, while the nonclassical form oscillates from 1:1000 to 1:3000 in males and 1:6000 to 1:40,000 in females. In this regard, patients with the classic phenotype have less than 1% of normal α-Gal A activity and tend to develop complications and symptoms earlier in life. Patients with the nonclassic subtype have a milder form of the disease, with higher α-Gal A activity [
233]. In contrast, although females are typically asymptomatic, a small percentage of them can exhibit a milder pattern of the disease due to continued secretion of α-Gal A from their other X chromosome.
Clinical features of FD include auditory disturbances, renal disease, cerebrovascular disease, cardiac hypertrophy, arrhythmia, angiokeratoma (skin lesions), and excessive sweating [
234]. From a molecular perspective, over 900 mutations have been identified in association with FD; D313Y, E66Q, and A143T are among the most common associated mutations [
235,
236].
In this scenario, taking into account the rest of the sphingolipidoses, in FD there is an increased basal expression of the autophagosome marker LC3-II, as observed in cultured cells from FD patients compared to wild-type cells. Furthermore, human podocytes derived from patients with FD have increased expression of GABARAP and BECN1 along with an inhibition of mTOR. However, an impairment in the autophagy flux in FD is described. According to this alteration, an increased staining of p62 and ubiquitin was observed in renal tissues and cultured fibroblasts from FD patients [
237]. Moreover, the accumulation of autophagy substrates, autophagosomes, and lysosomes was proved using an α-Gal A-deficient mouse model [
238]. These changes suggest that autophagy dysfunction could contribute to the progression of neuropathological changes in FD [
239] (
Figure 4).
Figure 4. Sphingolipidoses overview. Gaucher disease, Niemann–Pick type C disease, and Fabry disease. GD I, II and III correspond to the three different presentations of Gaucher Disease.
5. Autophagy Pathway in Neuronal Ceroid Lipofuscinoses
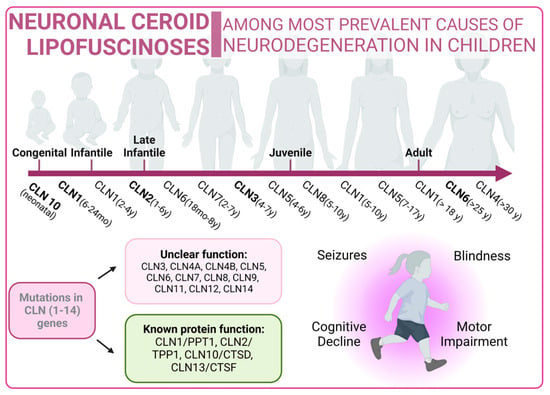
Neuronal ceroid lipofuscinoses (NCLs) are one of the most prevalent causes of neurodegeneration in children. NCLs typically present with symptoms such as blindness, seizures, progressive cognitive decline, and motor impairment. NCLs exhibit both genetic and phenotypic diversity. The juvenile onset NCL (JNCL), also known as Batten disease, is the most common presentation and is caused by mutations in the CLN3 gene [
240]. However, NCLs could manifest as different subtypes, each named according to the specific mutations in specific ceroid lipofuscinosis neuronal (CLN) genes such as CLN1, CLN2, CLN4, CLN5, CLN6, CLN7, CLN8, CLN10, CLN11, CLN12, or CLN13 [
241]. Mutations related to NCLs are listed in Supplementary Table S4 [
242,
243,
244,
245,
246,
247,
248,
249,
250,
251,
252,
253,
254,
255,
256,
257,
258,
259,
260,
261,
262,
263].
The predicted functions of CLN proteins are diverse, with some acting as lysosomal enzymes, while others are thought to regulate intracellular trafficking or membrane transport. Unfortunately, the precise cellular roles of most CLN proteins remain a mystery, prompting the utilization of a wide variety of model systems in NCL research. However, these diseases share the common features of autophagosome accumulation, dysfunctional mitochondria, and alterations in autophagy-related pathways, providing important insights into their pathogenesis [
264,
265].
Batten disease is an autosomal recessive disorder typically affecting children between the ages of 5 and 10 caused by mutations in CLN3. A frequently observed defect in CLN3 is the homozygous deletion of 966 base pairs, encompassing exons 7 and 8, resulting in a premature stop codon in exon 9 [
266]. In this context, the CLN3 protein is located in many cellular compartments, including the endo-lysosomal pathway and the Golgi complex. However, although its exact function is not fully understood, it has been linked to intracellular trafficking through interactions with Rab7A and protein secretion processes [
267,
268].
In this context, several mammalian models of NCLIII disease have shown reduced trafficking and levels of lysosomal enzymes. Early-stage defects in the autophagy pathway have been observed in both mouse and human cellular models with CLN3 mutations, leading to the accumulation of autophagosomes and autolysosomes. In particular, Cln3Dex7/8 knock-in mice and Cln3Dex7/8 cerebellar cells exhibit increased levels of LC3-II, downregulation of mTORC1, accumulation of autophagosomes, and impaired maturation as well as impaired turnover of ATP synthase subunit C [
269,
270].
On the other hand, the NCL X subtype (characterized by mutations in the Cathepsin D gene) also presents an increase in the number of autophagosomes and an accumulation of malfunctioning mitochondria due to an impairment in the autophagy flux [
271] (
Figure 5).
Figure 5. Neuronal Ceroid Lipofuscinoses overview. Classical forms of the disease are marked in bold type.
6. Autophagy in Glycoproteinoses
Glycoproteinoses form a category of lysosomal diseases resulting from deficiencies in the catabolism of glycoproteins. In this regard, glycoproteins are prevalent components found in cells and on cell surfaces. These genetic disorders follow an autosomal recessive inheritance pattern. Glycoproteinoses share the deficit of specific lysosomal enzymes that are crucial for the systematic breakdown of glycoprotein glucids [
272]. Pathogenic sequence variants in genes encoding these enzymes lead to glycoproteinoses. The oligosaccharide composition serves as an indicator of glycoproteinosis, potentially offering insights into a specific diagnosis [
273]. In this context, there are some deficiencies encompassed in this type of pathology, such as fucosidosis, galactosialidosis, Schindler’s disease, α-Mannosidosis, or β-Mannosidosis, among others [
274]. In many of these disorders, the accumulation of undigested material induces vacuolization in cells, such as peripheral blood cells and fibroblasts. This accumulation can also have pleiotropic effects on cellular functions, including synaptic release, exocytosis, and autophagy [
273]. Mutations related to glycoproteinoses are listed in Supplementary Table S5 [
275,
276,
277,
278,
279,
280,
281,
282,
283,
284,
285,
286,
287,
288,
289].
6.1. α-Mannosidosis
α-mannosidosis, is an uncommon LSD (1:500.000) and follows an autosomal recessive inheritance pattern. Typically, it is unnoticeable at birth and displays symptoms progressively [
290]. Clinical features of this disease include cognitive developmental delay, hearing loss, skeletal deformities, central nervous system involvement, and immunodeficiencies. Traditionally, α-mannosidosis is classified into two categories, according to its severity. However, wider classification includes three clinical types differentiated by the age of onset, speed of progression, and presence/absence of skeletal abnormalities. Type 1 includes mild presentation, an age of onset after 10 years old, no skeletal implication, and very slow progression. Type 2 is the moderate form, in which symptoms are identified before 10 years old, including skeletal abnormalities, with a slow progression that leads to ataxia around the age of 30. Type 3 is the most severe presentation and is immediately recognizable, exhibiting skeletal abnormalities. It progresses fast, culminating in early death due to central nervous system involvement or myopathy. The most frequent presentation is Type 2 [
291,
292,
293].
From a genetic point of view, α-mannosidosis is originated by MAN2B1 gene mutations, generating an aberrant enzyme (α-mannosidase) that is unable to break down mannose-containing oligosaccharides [
294]. Consequently, these accumulate within lysosomes, triggering cell malfunction and eventual cell death. The build-up of oligosaccharides and ensuing cell death contribute to tissue and organ damage, resulting in the distinctive features observed in α-mannosidosis [
295].
6.2. β-Mannosidosis
β-mannosidosis is an autosomal recessive LSD that occurs as a result of a dysfunctional β-mannosidase enzyme. While well-known and relatively common in goats and other types of cattle, it is exceptionally rare in humans. For this reason, the characterization of this pathology in a human context poses significant challenges due to its infrequency and limited occurrence in the human population [
296,
297].
The signs and symptoms of β-mannosidosis present a wide spectrum of severity, and onset can occur from infancy to adulthood. In fact, there is no clear genotype/phenotype correlation. Common symptoms include intellectual disability, and some may experience delayed motor development and seizures. Behavioral problems, such as hyperactivity, impulsivity, aggressiveness, or a tendency to depression; respiratory and ear infections; hearing loss; speech impairment; swallowing difficulties; and hypotonia, are additional challenges faced by those with β-mannosidosis and are also common among patients who may exhibit introverted behavior [
298,
299]. Distinctive facial features and the presence of small angiokeratomas, formed by clusters of enlarged blood vessels, are also observed in individuals with this condition.
From a molecular point of view, β-mannosidosis is caused by alterations in the MANBA gene [
300]. These disrupt the function of the β-mannosidase enzyme, which serves as the final exoglycosidase in the degradation of N-linked oligosaccharides of glycoproteins, removing β-linked mannose residues [
301]. Affected individuals exhibit a significant reduction in β-mannosidase activity, and this leads to the lysosomal accumulation of disaccharides.
This entry is adapted from the peer-reviewed paper 10.3390/biology13010034