 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francesca Lofrumento | -- | 2723 | 2023-12-25 11:39:02 | | | |
| 2 | Jessie Wu | + 2 word(s) | 2725 | 2023-12-26 02:42:25 | | | | |
| 3 | Jessie Wu | -2 word(s) | 2723 | 2024-02-28 09:53:24 | | |
Video Upload Options
Ischemic heart disease holds the foremost position as the primary contributor to mortality from cardiovascular disease (CVD). Furthermore, it constitutes the predominant underlying cause of heart failure on a global scale. Diverging from other tissues, the myocardium demonstrates a markedly limited ability to regenerate in the aftermath of injuries. Consequently, necrotic cardiomyocytes are replaced by fibrotic scar tissue in the cardiac repair process, which can lead to an adverse cardiac remodeling. Different cell types, including fibroblasts and macrophages, are involved in this process and play a pivotal role by releasing a wide array of mediators (i.e., cytokines) that regulate the activation of multiple molecular pathways, such as the Wnt/β-catenin pathway, involved in cardiac fibrosis. For this reason, the modulation of these pathways might be effective in promoting the replacement of fibrosis in reactive tissue. Dual antiplatelet therapy (DAPT), consisting of the combination of a platelet P2Y12 receptor inhibitor and aspirin, is the cornerstone of treatment for patients with acute coronary syndromes (ACS) requiring percutaneous coronary interventions (PCI). P2Y12 receptor activation, a platelet purinergic receptor for adenosine 5′-diphosphate (ADP), significantly contributes to the arterial thrombosis process.
1. P2Y12 Platelet Receptors and Inhibitors
2. Non-Platelet P2Y12 Receptors
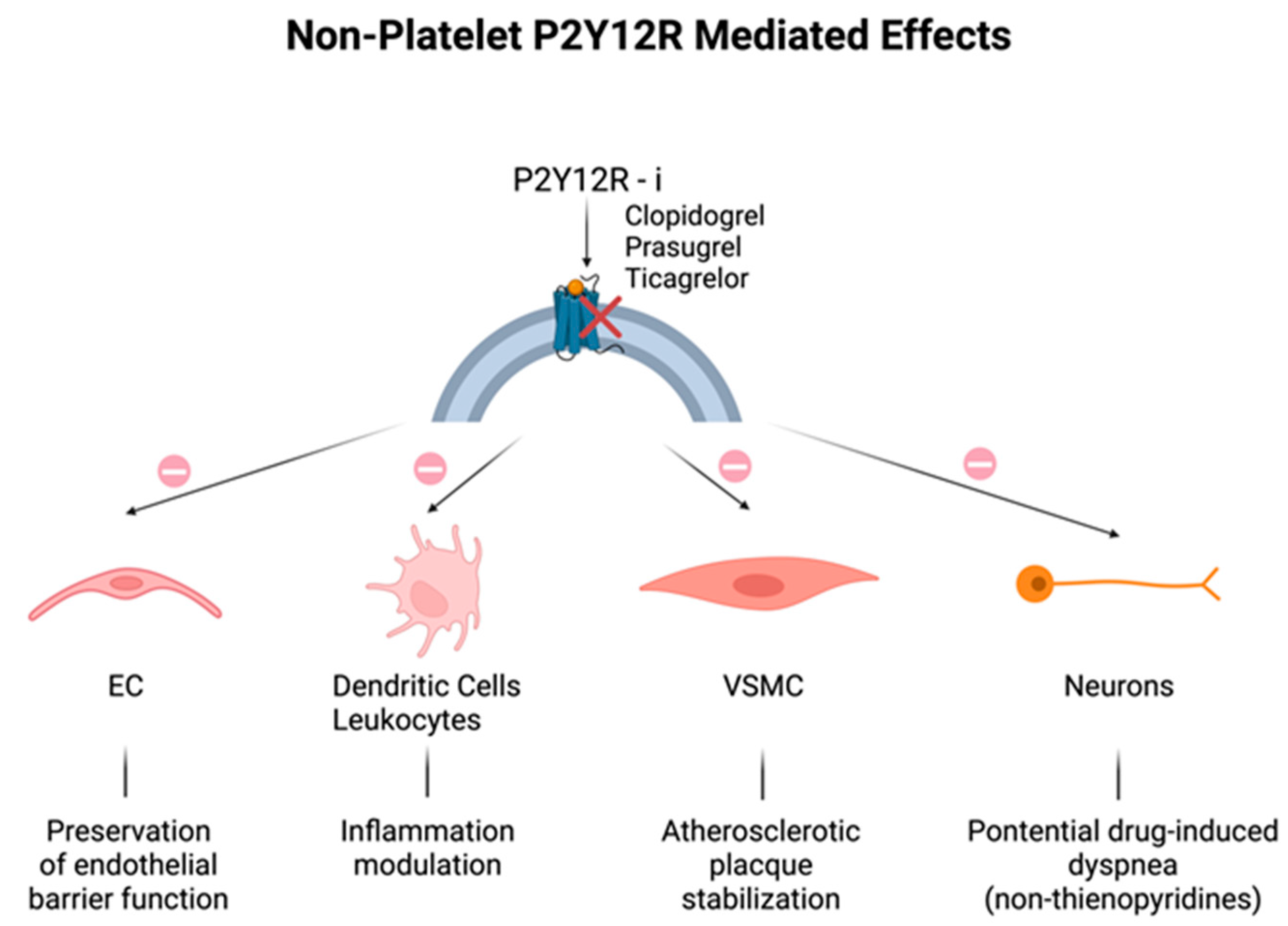
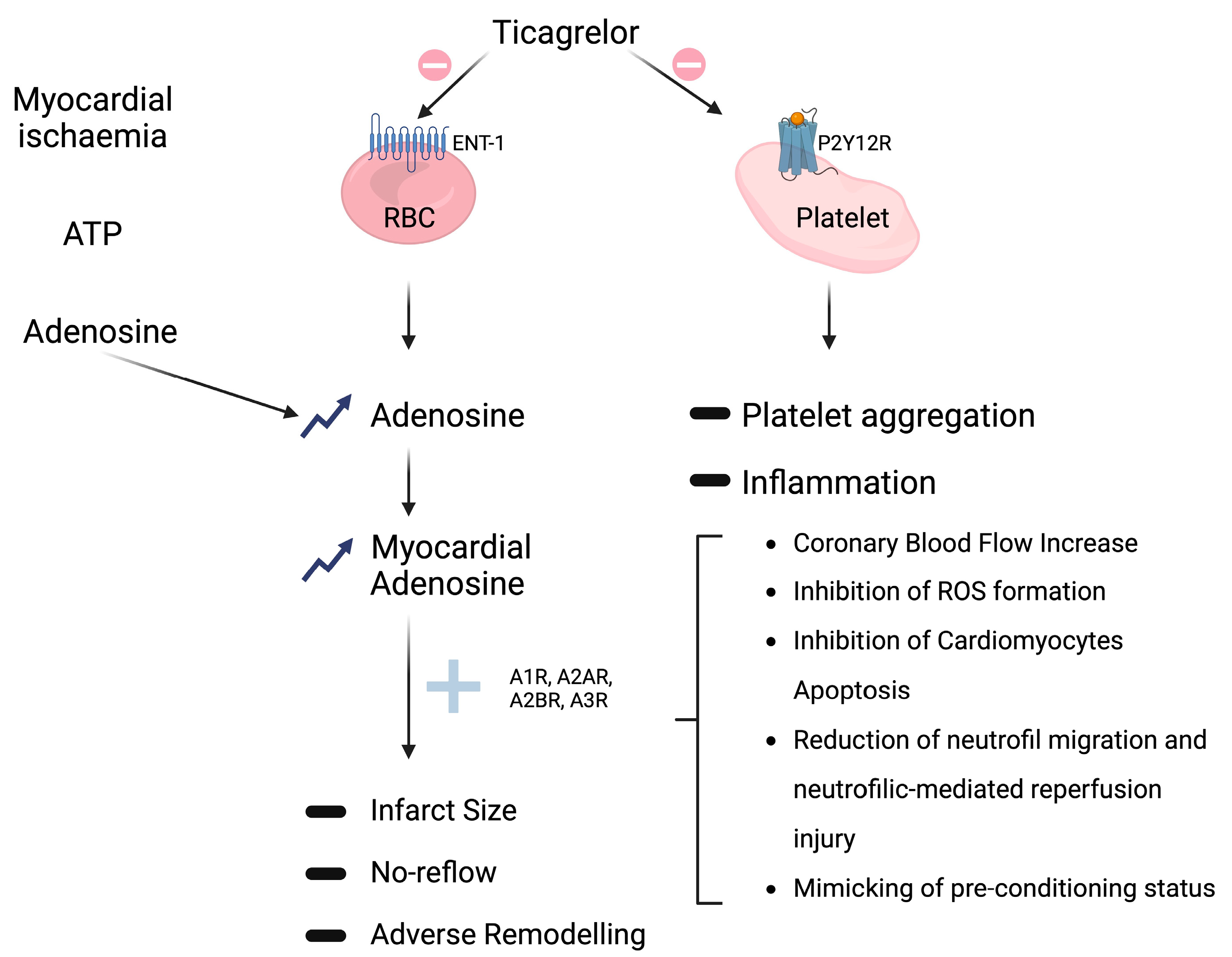
3. Non-P2Y12-Mediated Effects
Adenosine-Mediated Effects
References
- Valgimigli, M.; Bueno, H.; Byrne, R.; Collet, J.-P.; Costa, F.; Jeppsson, A.; Jüni, P.; Kastrati, A.; Kolh, P.; Mauri, L.; et al. 2017 ESC focused update on dual antiplatelet therapy in coronary artery disease developed in collaboration with EACTS. Eur. Heart J. 2018, 39, 213–260, Corrigendum in Eur. Heart J. 2018, 39, 2089.
- Adamski, P.; Koziński, M.; Ostrowska, M.; Fabiszak, T.; Navarese, E.P.; Paciorek, P.; Grześk, G.; Kubica, J. Overview of pleiotropic effects of platelet P2Y12 receptor inhibitors. Thromb. Haemost. 2014, 112, 224–242.
- Nylander, S.; Schulz, R. Effects of P2Y12 receptor antagonists beyond platelet inhibition–comparison of ticagrelor with thienopyridines. Br. J. Pharmacol. 2016, 173, 1163–1178.
- Huang, Z.; Xie, N.; Illes, P.; Di Virgilio, F.; Ulrich, H.; Semyanov, A.; Verkhratsky, A.; Sperlagh, B.; Yu, S.-G.; Huang, C.; et al. From purines to purinergic signalling: Molecular functions and human diseases. Signal Transduct. Target. Ther. 2021, 6, 162.
- Mansour, A.; Bachelot-Loza, C.; Nesseler, N.; Gaussem, P.; Gouin-Thibault, I. P2Y12 Inhibition beyond Thrombosis: Effects on Inflammation. Int. J. Mol. Sci. 2020, 21, 1391.
- Cattaneo, M.; Schulz, R.; Nylander, S. Adenosine-Mediated Effects of Ticagrelor. J. Am. Coll. Cardiol. 2014, 63, 2503–2509.
- Wallentin, L. P2Y12 inhibitors: Differences in properties and mechanisms of action and potential consequences for clinical use. Eur. Heart J. 2009, 30, 1964–1977.
- Byrne, R.A.; Rossello, X.; Coughlan, J.J.; Barbato, E.; Berry, C.; Chieffo, A.; Claeys, M.J.; Dan, G.-A.; Dweck, M.R.; Galbraith, M.; et al. 2023 ESC Guidelines for the management of acute coronary syndromes. Eur. Heart J. 2023, 44, 3720–3826.
- Procopio, M.C.; Lauro, R.; Nasso, C.; Carerj, S.; Squadrito, F.; Bitto, A.; Di Bella, G.; Micari, A.; Irrera, N.; Costa, F. Role of Adenosine and Purinergic Receptors in Myocardial Infarction: Focus on Different Signal Transduction Pathways. Biomedicines 2021, 9, 204.
- Baqi, Y.; Müller, C.E. Antithrombotic P2Y12 receptor antagonists: Recent developments in drug discovery. Drug Discov. Today 2019, 24, 325–333.
- Aslam, M.; Tanislav, C.; Troidl, C.; Schulz, R.; Hamm, C.; Gündüz, D. cAMP controls the restoration of endothelial barrier function after thrombin-induced hyperpermeability via Rac1 activation. Physiol. Rep. 2014, 2, e12175.
- Teng, R.; Oliver, S.; Hayes, M.A.; Butler, K. Absorption, Distribution, Metabolism, and Excretion of Ticagrelor in Healthy Subjects. Drug Metab. Dispos. 2010, 38, 1514–1521.
- Wallentin, L.; Becker, R.C.; Budaj, A.; Cannon, C.P.; Emanuelsson, H.; Held, C.; Horrow, J.; Husted, S.; James, S.; Katus, H.; et al. Ticagrelor versus Clopidogrel in Patients with Acute Coronary Syndromes. N. Engl. J. Med. 2009, 361, 1045–1057.
- Wiviott, S.D.; Braunwald, E.; McCabe, C.H.; Montalescot, G.; Ruzyllo, W.; Gottlieb, S.; Neumann, F.-J.; Ardissino, D.; De Servi, S.; Murphy, S.A.; et al. Prasugrel versus Clopidogrel in Patients with Acute Coronary Syndromes. N. Engl. J. Med. 2007, 357, 2001–2015.
- Aradi, D.; Kirtane, A.; Bonello, L.; Gurbel, P.A.; Tantry, U.S.; Huber, K.; Freynhofer, M.K.; ten Berg, J.; Janssen, P.; Angiolillo, D.J.; et al. Bleeding and stent thrombosis on P2Y12-inhibitors: Collaborative analysis on the role of platelet reactivity for risk stratification after percutaneous coronary intervention. Eur. Heart J. 2015, 36, 1762–1771.
- Husted, S.; James, S.; Becker, R.C.; Horrow, J.; Katus, H.; Storey, R.F.; Cannon, C.P.; Heras, M.; Lopes, R.D.; Morais, J.; et al. Ticagrelor Versus Clopidogrel in Elderly Patients with Acute Coronary Syndromes. Circ. Cardiovasc. Qual. Outcomes 2012, 5, 680–688.
- Hechler, B.; Gachet, C. Purinergic Receptors in Thrombosis and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2307–2315.
- Thomas, M.R.; Storey, R.F. The role of platelets in inflammation. Thromb. Haemost. 2015, 114, 449–458.
- Aslam, M.; Rohrbach, S.; Rafiq, A.; Nazli, S.; Piper, H.M.; Noll, T.; Schulz, R.; Gündüz, D.; Schluter, K.-D. Hypoxia-reoxygenation-induced endothelial barrier failure: Role of RhoA, Rac1 and myosin light chain kinase. J. Physiol. 2013, 591, 461–473.
- Satonaka, H.; Nagata, D.; Takahashi, M.; Kiyosue, A.; Myojo, M.; Fujita, D.; Ishimitsu, T.; Nagano, T.; Nagai, R.; Hirata, Y. Involvement of P2Y12 receptor in vascular smooth muscle inflammatory changes via MCP-1 upregulation and monocyte adhesion. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H853–H861.
- Cattaneo, M.; Faioni, E.M. Why does ticagrelor induce dyspnea? Thromb. Haemost. 2012, 108, 1031–1036.
- van Giezen, J.J.J.; Sidaway, J.; Glaves, P.; Kirk, I.; Björkman, J.-A. Ticagrelor Inhibits Adenosine Uptake In Vitro and Enhances Adenosine-Mediated Hyperemia Responses in a Canine Model. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 164–172.
- Nanhwan, M.K.; Ling, S.; Kodakandla, M.; Nylander, S.; Ye, Y.; Birnbaum, Y. Chronic Treatment with Ticagrelor Limits Myocardial Infarct Size. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2078–2085.
- Gurbel, P.A.; Jeong, Y.-H.; Tantry, U.S. The Dogged Search for Cryptic Effects of Ticagrelor. Circulation 2016, 134, 1720–1723.
- Vilahur, G.; Gutiérrez, M.; Casani, L.; Varela, L.; Capdevila, A.; Pons-Lladó, G.; Carreras, F.; Carlsson, L.; Hidalgo, A.; Badimon, L.; et al. Protective Effects of Ticagrelor on Myocardial Injury After Infarction. Circulation 2016, 134, 1708–1719.
- Alexopoulos, D.; Moulias, A.; Koutsogiannis, N.; Xanthopoulou, I.; Kakkavas, A.; Mavronasiou, E.; Davlouros, P.; Hahalis, G.; R, O.; W, P.; et al. Differential Effect of Ticagrelor Versus Prasugrel on Coronary Blood Flow Velocity in Patients with Non–ST-Elevation Acute Coronary Syndrome Undergoing Percutaneous Coronary Intervention. Circ. Cardiovasc. Interv. 2013, 6, 277–283.
- Bonello, L.; Laine, M.; Kipson, N.; Mancini, J.; Helal, O.; Fromonot, J.; Gariboldi, V.; Condo, J.; Thuny, F.; Frere, C.; et al. Ticagrelor Increases Adenosine Plasma Concentration in Patients with an Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2014, 63, 872–877.
- Wittfeldt, A.; Emanuelsson, H.; Brandrup-Wognsen, G.; van Giezen, J.; Jonasson, J.; Nylander, S.; Gan, L.-M. Ticagrelor Enhances Adenosine-Induced Coronary Vasodilatory Responses in Humans. J. Am. Coll. Cardiol. 2013, 61, 723–727.
- Jeong, H.S.; Hong, S.J.; Cho, S.-A.; Kim, J.-H.; Cho, J.Y.; Lee, S.H.; Joo, H.J.; Park, J.H.; Yu, C.W.; Lim, D.-S. Comparison of Ticagrelor Versus Prasugrel for Inflammation, Vascular Function, and Circulating Endothelial Progenitor Cells in Diabetic Patients with Non–ST-Segment Elevation Acute Coronary Syndrome Requiring Coronary Stenting. JACC Cardiovasc. Interv. 2017, 10, 1646–1658.
- Ariotti, S.; Ortega-Paz, L.; van Leeuwen, M.; Brugaletta, S.; Leonardi, S.; Akkerhuis, K.M.; Rimoldi, S.F.; Janssens, G.; Gianni, U.; Berge, J.C.v.D.; et al. Effects of Ticagrelor, Prasugrel, or Clopidogrel on Endothelial Function and Other Vascular Biomarkers. JACC Cardiovasc. Interv. 2018, 11, 1576–1586.
- Storey, R.F.; James, S.K.; Siegbahn, A.; Varenhorst, C.; Held, C.; Ycas, J.; Husted, S.E.; Cannon, C.P.; Becker, R.C.; Steg, P.G.; et al. Lower mortality following pulmonary adverse events and sepsis with ticagrelor compared to clopidogrel in the PLATO study. Platelets 2014, 25, 517–525.
- Alsharif, K.F.; Thomas, M.R.; Judge, H.M.; Khan, H.; Prince, L.R.; Sabroe, I.; Ridger, V.C.; Storey, R.F. Ticagrelor potentiates adenosine-induced stimulation of neutrophil chemotaxis and phagocytosis. Vasc. Pharmacol. 2015, 71, 201–207.
- Bonaca, M.P.; Bhatt, D.L.; Cohen, M.; Steg, P.G.; Storey, R.F.; Jensen, E.C.; Magnani, G.; Bansilal, S.; Fish, M.P.; Im, K.; et al. Long-Term Use of Ticagrelor in Patients with Prior Myocardial Infarction. N. Engl. J. Med. 2015, 372, 1791–1800.
- Sexton, T.R.; Zhang, G.; Macaulay, T.E.; Callahan, L.A.; Charnigo, R.; Vsevolozhskaya, O.A.; Li, Z.; Smyth, S. Ticagrelor Reduces Thromboinflammatory Markers in Patients with Pneumonia. JACC Basic. Transl. Sci. 2018, 3, 435–449.
- Lancellotti, P.; Musumeci, L.; Jacques, N.; Servais, L.; Goffin, E.; Pirotte, B.; Oury, C. Antibacterial Activity of Ticagrelor in Conventional Antiplatelet Dosages Against Antibiotic-Resistant Gram-Positive Bacteria. JAMA Cardiol. 2019, 4, 596–599.
- Armstrong, D.; Summers, C.; Ewart, L.; Nylander, S.; Sidaway, J.E.; van Giezen, J.J.J. Characterization of the Adenosine Pharmacology of Ticagrelor Reveals Therapeutically Relevant Inhibition of Equilibrative Nucleoside Transporter 1. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 209–219.
- Babbitt, D.G.; Virmani, R.; Forman, M.B. Intracoronary adenosine administered after reperfusion limits vascular injury after prolonged ischemia in the canine model. Circulation 1989, 80, 1388–1399.
- Thornton, J.D.; Liu, G.S.; Olsson, R.A.; Downey, J.M. Intravenous pretreatment with A1-selective adenosine analogues protects the heart against infarction. Circulation 1992, 85, 659–665.
- Tantry, U.S.; Jeong, Y.-H.; Gurbel, P.A. More Evidence for Non-P2Y12-Mediated Effects of Ticagrelor. JACC Cardiovasc. Interv. 2017, 10, 1659–1661.
- Tao, L.; Ren, S.; Zhang, L.; Liu, W.; Zhao, Y.; Chen, C.; Mao, X.; Chen, Z.; Gu, X. A Review of the Role of the Antiplatelet Drug Ticagrelor in the Management of Acute Coronary Syndrome, Acute Thrombotic Disease, and Other Diseases. Med. Sci. Monit. 2022, 28, e935664.
- da Silva, J.S.; Gabriel-Costa, D.; Sudo, R.T.; Wang, H.; Groban, L.; Ferraz, E.B.; Nascimento, J.H.M.; Fraga, C.A.M.; Barreiro, E.J.; Zapata-Sudo, G. Adenosine A2A receptor agonist prevents cardiac remodeling and dysfunction in spontaneously hypertensive male rats after myocardial infarction. Drug Des. Devel Ther. 2017, 11, 553–562.
- da Silva, J.S.; Pereira, S.L.; Maia, R.D.C.; Landgraf, S.S.; Caruso-Neves, C.; Kümmerle, A.E.; Fraga, C.A.M.; Barreiro, E.J.; Sudo, R.T.; Zapata-Sudo, G. N-acylhydrazone improves exercise intolerance in rats submitted to myocardial infarction by the recovery of calcium homeostasis in skeletal muscle. Life Sci. 2014, 94, 30–36.
- Villarreal, F.; Epperson, S.A.; Ramirez-Sanchez, I.; Yamazaki, K.G.; Brunton, L.L. Regulation of cardiac fibroblast collagen synthesis by adenosine: Roles for Epac and PI3K. Am. J. Physiol. Cell Physiol. 2009, 296, C1178–C1184.
- Guo, F.; Wang, X.; Guo, Y.; Wan, W.; Cui, Y.; Wang, J.; Liu, W. Shenfu Administration Improves Cardiac Fibrosis in Rats with Myocardial Ischemia-Reperfusion Through Adenosine A2a Receptor Activation. Hum. Exp. Toxicol. 2022, 41, 096032712210776.
- Tamargo, J.; López-Sendón, J. Novel therapeutic targets for the treatment of heart failure. Nat. Rev. Drug Discov. 2011, 10, 536–555.

