1. P2Y12 Platelet Receptors and Inhibitors
Among extracellular nucleotides, ADP assumes a central role in platelet function and the initiation of thrombus formation. Platelets express two purinergic receptors for ADP, namely P2Y1 and P2Y12, which are coupled to Gq (G-protein q) and Gi (G-protein i), respectively [
7,
8,
9]. Of these, P2Y12 holds particular significance. Stimulation of P2Y1 by ADP leads to the activation of the glycoprotein (GP) IIb/IIIa receptor, resulting in calcium mobilization, alterations in platelet shape, and transient platelet aggregation. In contrast, activation of P2Y12 triggers the GP IIb/IIIa receptor, culminating in platelet degranulation, thromboxane production, and prolonged platelet aggregation. This, in turn, establishes a positive feedback loop that contributes significantly to the formation of arterial thrombi [
12,
13]. This thrombotic process represents a hallmark of myocardial infarction (MI) pathogenesis, underscoring the identification of P2Y12 as a therapeutic target for the management and prevention of arterial thrombosis [
7].
Ticlopidine was the first P2Y12 antagonist approved by the Food and Drug Administration (FDA) in 1991 [
7]. Subsequently, several P2Y12 inhibitors have been introduced into clinical practice. These P2Y12 inhibitors are categorized into two classes of drug: the thienopyridines, comprising clopidogrel, prasugrel, and ticlopidine, and the non-thienopyridine derivatives such as ticagrelor, cangrelor, and elinogrel. Thienopyridines are administered orally as prodrugs and undergo hepatic and intestinal metabolism to yield active metabolites, which subsequently bind covalently to the P2Y12 receptor, thereby inducing irreversible platelet inhibition throughout their lifespan. The production of these active metabolites, particularly in the case of clopidogrel, relies on cytochrome P450 (CYP450) enzymes in the liver. However, clopidogrel is accompanied by several limitations, including a delayed onset of action, variability in response, resistance to the drug, moderate inhibition of platelet aggregation (ranging from 33% to 64%), and potential drug interactions due to the involvement of CYP450 enzymes and genetic variations [
11,
12]. In response to these challenges, the third-generation thienopyridine, prasugrel, was developed. In contrast to ticlopidine and clopidogrel, prasugrel initially undergoes metabolism by an intestinal esterase. This initial step is followed by a single-step activation process dependent on CYP450 enzymes. This distinctive metabolic pathway results in a swifter onset of action, more potent inhibition of platelet aggregation (ranging from 50% to 80%), reduced resistance to the drug, and a lower likelihood of drug interactions. However, it is worth noting that prasugrel is associated with an increased risk of bleeding [
11,
12]. The second category, consisting of non-thienopyridine derivatives, does not necessitate hepatic bioactivation and delivers reversible P2Y12 receptor inhibition [
6]. Among these, ticagrelor stands out as an oral direct-acting P2Y12 inhibitor. It boasts a rapid onset of action within two hours and exerts a substantial inhibitory effect on platelet aggregation, ranging from 60% to 90%. Additionally, the active metabolite, AR–C124910XX, is generated through hepatic metabolism of ticagrelor and contributes to one-third of its antiplatelet effects. Based on the findings of the phase III Platelet Inhibition and Patient Outcomes (PLATO) trial and the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel Thrombolysis In Myocardial Infarction 38 (TRITON-TIMI 38), it is recommended, as the primary strategy, to employ dual antiplatelet therapy (DAPT) comprising both aspirin and a potent P2Y12 receptor inhibitor, such as prasugrel or ticagrelor, for patients with acute coronary syndrome (ACS) [
5,
14,
15]. The use of clopidogrel, which offers less effective and more variable platelet inhibition, should be limited to situations where prasugrel or ticagrelor are contraindicated or unavailable, or in specific instances where there is a high risk of bleeding (according to criteria such as ≥1 major or ≥2 minor ARC-HBR criteria) [
10,
16]. Additionally, the consideration of clopidogrel may be appropriate for elderly patients (i.e., those aged ≥ 70 years) [
5,
10,
17].
2. Non-Platelet P2Y12 Receptors
In addition to their established hemostatic functions, platelets are increasingly recognized for their substantial involvement in modulating inflammatory and immune responses. This recognition is attributable to their interaction with immune cells through the exposure of membrane proteins such as P-selectin and CD40 ligand (cluster of differentiation 40), as well as their release of inflammatory mediators, including cytokines and chemokines [
8,
18,
19].
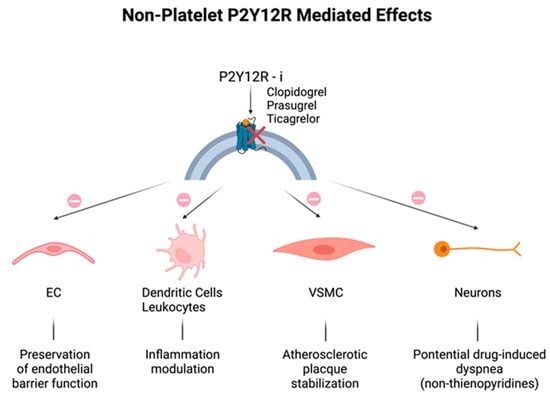
Historically, P2Y12 receptors have been recognized to be specific in platelets, but the discovery of their expression in other cells, including endothelial cells (ECs), vascular smooth muscle cells (VSMCs), immune cells, dendritic cells, and neurons could explain the additional effects related to their modulation (
Figure 1) [
8,
20,
21]. As for ECs, disruption of endothelial barrier function was attenuated by the increase in EC cyclic adenosine monophosphate (cAMP) levels, because of the P2Y12 receptor blockade [
22,
23]. P2Y12 receptor activation on VSMCs may induce an inflammatory state closely associated with atherosclerotic plaque instability [
24], that may be mitigated by P2Y12 receptor inhibitors. Lastly, neuronal P2Y12 receptors might seem to be the mediators of drug-induced dyspnea observed in patients treated with non-thienopyridine derivatives [
25]; in fact, repeated dosing of reversible P2Y12 receptor inhibitors, like ticagrelor, ensures a continuous blockage of neural P2Y12 receptors, in contrast to temporary and transient clopidogrel interaction, due to the fact that, unlike platelets, nucleated cells exhibit the capacity to promptly substitute the suppressed receptors [
11].
Figure 1. Non-platelet P2Y12 receptor (P2Y12R)-mediated off-target effects: additional effects of all P2Y12R inhibitors on different cell types. (Top): P2Y12 receptor blockade (red cross) by clopidogrel, prasugrel, and ticagrelor. (Bottom): effects (arrows) of PY12R inhibition (prohibition symbol) on endothelial cells (ECs), immune cells, vascular smooth muscle cells (VSMCs), and neurons.
Therefore, the impact and clinical advantages of P2Y12 receptor antagonism may extend beyond merely platelet inhibition and arterial thrombosis prevention. Additional potential consequences encompass the attenuation of the pro-inflammatory effects of activated platelets (
Figure 2) and the impact of P2Y12 receptor inhibition on various other cell populations [
8,
20,
23].
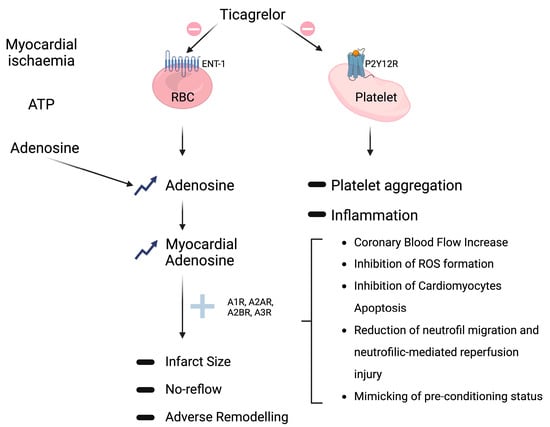
Figure 2. Non-P2Y12-mediated off-target effects of ticagrelor. (
Top right): conventional platelet PY12R inhibition (prohibition symbol) resulting (arrows) in reduction (minus sign) of platelet aggregation and inflammation (see
Section 2.2). (
Middle): ticagrelor off-target inhibition (prohibition symbol) of adenosine reuptake via sodium-independent equilibrative nucleoside transporters 1 (ENT1) on red blood cells (RBCs) that leads (arrows) to both systemic and tissue adenosine levels elevation (upward arrows); adenosine interaction (plus sign) with its receptors (A1R, A2AR, A2BR, and A3R) culminates, through different possible mechanisms (square bracket), in multiple favorable outcomes, like reduction (minus sign) of infarct size, no-reflow, and adverse remodeling. (
Top left): adenosine production (arrows) from adenosine triphosphate (ATP) degradation by CD39 and CD73 enzymes in response to hypoxia and tissue damage.
3. Non-P2Y12-Mediated Effects
In the PLATO (Platelet Inhibition and Patient Outcomes) trial, ticagrelor demonstrated superiority over clopidogrel in prevention of cardiovascular death, myocardial infarction, or stroke, with incidence rates of 9.8% compared to 11.7%, resulting in a relative risk reduction of 16% [
18]. An additional positive effect on all-cause mortality, which was reduced by 24% by ticagrelor compared to clopidogrel, has been observed. Considering that such mortality benefit was not observed in the pivotal trial of prasugrel, a potent P2Y12 inhibitor with a comparable P2Y12 inhibitory effect, such mortality benefit has been speculated to be linked to the additional non-P2Y12-mediated off-target mechanisms of the drug [
19].
The most extensively documented off-target mechanism pertains to ticagrelor’s inhibition of adenosine reuptake by red blood cells, accomplished via sodium-independent equilibrative nucleoside transporter 1 (ENT1). This action leads to the elevation of both systemic and tissue adenosine levels (
Figure 2) [
6,
20,
23]. Van Giezen et al. illustrated that ticagrelor inhibits adenosine reuptake in erythrocytes, resulting in an elevation of adenosine levels in the bloodstream, accompanied by a notable, dose-dependent enhancement of adenosine-mediated coronary blood flow [
26]. Furthermore, ticagrelor was also able to reduce the size of the infarction area, an effect not observed following clopidogrel treatment [
27]. Vilahur et al. provide additional evidence supporting the non-P2Y12-mediated hypothesis—ticagrelor significantly reduced the infarct size compared to clopidogrel—an effect substantiated through the evaluation of troponin-I levels and histopathological analysis. Moreover, in comparison to clopidogrel, ticagrelor led to a significant reduction in myocardial edema and activated both AMP-activated protein kinase signaling and cyclooxygenase-2. Importantly, the advantageous outcomes associated with ticagrelor in comparison to clopidogrel were hindered by the A1/A2 receptor antagonist 8-p-sulfophenyl theophylline. Therefore, these studies underline that ticagrelor may play an important cardioprotective role by decreasing necrotic injury and edema via adenosine-dependent mechanisms in addition to its antiplatelet role [
28,
29]. Regarding human investigations, it was observed that the maintenance dose of ticagrelor led to an augmentation of adenosine-induced coronary blood flow velocity in non-ST-elevation ACS patients undergoing percutaneous coronary intervention (PCI) [
30]. Furthermore, Bonello et al. provided evidence of ticagrelor significantly elevating adenosine plasma levels in comparison with clopidogrel in ACS patients. In the same study, it was noted that serum from patients treated with ticagrelor, but not clopidogrel, exhibited the capacity to inhibit the uptake of exogenous adenosine by erythrocytes in vitro [
31]. A double-blind, placebo-controlled, randomized study involving 40 healthy male subjects also showed that a single 180 mg loading dose of ticagrelor increased adenosine-induced coronary blood flow velocity. These beneficial effects, as well as the higher rate of dyspnea and bradycardia observed following ticagrelor administration, were probably related to higher adenosine levels, which were reversed by theophylline, an adenosine receptor antagonist, thus suggesting that all these effects are mediated by adenosine receptors [
32].
These findings affirm that oral administration of ticagrelor may be sufficient to impede the cellular uptake of adenosine, thus extending its half-life and elevating its concentration, thereby enhancing its cardioprotective effects. This off-target effect associated with ticagrelor has the potential to ameliorate endothelial function, although it should be noted that the current body of evidence remains somewhat limited [
6,
33]. In light of these considerations, the HI-TECH (Hunting for the Off-target Properties of Ticagrelor on Endothelial Function and Other Circulating Biomarkers in Humans) trial was undertaken to investigate the off-target effects of ticagrelor on endothelial function. This trial marked the third randomized study with this specific objective but was the first to juxtapose ticagrelor with both prasugrel and clopidogrel in a chronic setting, focusing on stabilized post-ACS patients. This approach was adopted to mitigate the potential confounding influences of the natural course of the disease, including the acute inflammatory phase of ACS and tissue ischemia, on the comparison across P2Y12 inhibitors and systemic adenosine plasma levels. In the context of this trial, assessments of endothelial-dependent dilatation, as gauged by the reactive hyperemia index (RHI), and, in a subgroup of patients, flow-mediated dilatation (FMD) of the brachial artery, showed no significant disparities between ticagrelor and prasugrel or clopidogrel at the presently approved regimens. Likewise, systemic adenosine plasma levels and vascular biomarkers exhibited no noteworthy distinctions at any of the time points measured [
34]. Conversely, a post hoc analysis of the PLATO trial unveiled a notable association between ticagrelor, in comparison with clopidogrel, and reduced morbidity and mortality attributable to pulmonary infection and sepsis [
35]. These observations stem from the combined effect of adenosine, which plays an augmenting role in modulating the inflammatory response, and the anti-inflammatory impact of P2Y12 receptor inhibition, as explained earlier. The potential of ticagrelor to confer anti-inflammatory effects through adenosine was also demonstrated in vitro, where ticagrelor was found to enhance adenosine-induced neutrophil migration [
36]. However, this beneficial effect did not manifest in the PEGASUS trial, where there was a numerical increase in deaths attributed to infections in the 90 mg ticagrelor group when compared to the 60 mg ticagrelor and placebo groups [
37]. It is important to note that these divergent outcomes between the PLATO and PEGASUS trials may be attributed to the distinct patient populations under study. For instance, the anti-inflammatory effect might have held particular significance for patients who had undergone coronary artery bypass grafts (CABGs), as they might have been more vulnerable to infections. In the PLATO trial, ticagrelor demonstrated a 50% reduction in mortality compared to clopidogrel in CABG patients [
35]. In the PEGASUS trial, which included a more stable patient cohort, fewer individuals would have undergone CABG, potentially explaining the differences in infection or sepsis-related mortality. More recently, the Targeting Platelet–Leukocyte Aggregates in Pneumonia with Ticagrelor (XANTHIPPE) study reported an association between ticagrelor and improved lung function in patients hospitalized for pneumonia [
38]. Notably, ticagrelor’s antimicrobial activity was recently established in mouse models, although it differs from the pharmacokinetics observed in humans [
39]. Therefore, further investigations are needed. In conclusion, a debate about the potential off-target adenosine-mediated effects of ticagrelor is still ongoing.
Importantly, it is noteworthy that only ticagrelor, and not its metabolite AR-C124910XX, exhibits a substantial capacity for inhibiting ENT1 [
40]. Additionally, aside from its ENT1 inhibitory effects, ticagrelor has demonstrated the induction of ATP release from human erythrocytes in vitro; however, the translation of this phenomenon to in vivo settings remains uncertain. Both of these mechanisms would act synergistically to further elevate adenosine concentration—ATP undergoes rapid conversion into AMP, facilitated by ectonucleoside triphosphate diphosphohydrolase 1 (CD39), ultimately leading to adenosine production via the action of ecto-5′-nucleotidase (CD73), as previously emphasized by Cattaneo et al. [
11].
Adenosine-Mediated Effects
Adenosine is locally generated in response to hypoxia and tissue damage, a process involving the degradation of adenosine triphosphate (ATP) and adenosine diphosphate (ADP) by CD39 and CD73 enzymes. Subsequently, adenosine is swiftly internalized through ENT1. The inhibition of ENT1 by ticagrelor leads to an intensified response to adenosine, which acts through interactions with adenosine receptors belonging to the G-protein-coupled receptor family, including (i) A1R and A3R, coupled to the inhibitory G protein (Gi), which inhibits adenylyl cyclase, thereby reducing intracellular cAMP, and (ii) A2AR and A2BR, coupled to the stimulatory G protein (Gs), which activates adenylyl cyclase, resulting in increased intracellular cAMP levels [
11,
14]. Pre-clinical investigations have reported multiple favorable outcomes associated with the use of adenosine agonists, positioning adenosine as a potential cardioprotective agent during acute myocardial infarction (AMI) (as depicted in
Figure 2) [
9,
41,
42]. Specifically, an increase in myocardial adenosine levels has been linked to augmented coronary blood flow, reduced expression of inflammatory markers, diminished fibrosis, reduced infarct size, decreased edema, and enhanced tissue remodeling [
6,
9,
43]. Furthermore, adenosine has demonstrated the ability to attenuate and potentiate ischemic preconditioning, thereby conferring protection against subsequent reperfusion injury [
9,
44].
Da Silva and colleagues have made a pioneering contribution by showcasing the effectiveness of orally administered LASSBio-294, an A2A-adenosine receptor agonist, in preventing the advancement of left ventricular (LV) dysfunction following myocardial infarction (MI) within a preclinical model characterized by preexisting hypertension [
45]. Notably, the same authors had previously established the efficacy of this compound in preventing heart failure in normotensive animals when administered intraperitoneally [
46]. LASSBio-294 effectively mitigated fibrotic and inflammatory responses, by reducing LV collagen deposition and tumor necrosis factor α expression [
45]. This discovery points to the potential role of cAMP-dependent signaling, regulated via A2AR activation, in modulating cardiac remodeling and the progression of heart failure by mitigating fibroblast proliferation and collagen synthesis [
45,
47]. Additionally, a study conducted in China in 2022 shed light on the beneficial effects of Shenfu injection (SFI), a commonly employed treatment for cardiac dysfunction in China. The study illustrated that SFI attenuated cardiac fibrosis through the activation of adenosine A2A receptors (A2ARs) in a rat model of myocardial ischemia-reperfusion (MI/R) [
48].
Therefore, the clinical use of A2A receptor agonists could be a potential alternative treatment for heart failure, in addition to the conventional anti-remodeling therapies. In fact, considering the modest efficacy of traditional therapies in terms of cardiovascular morbidity and mortality [
49], it is worth exploring new pharmacological targets for heart-failure treatment.
This entry is adapted from the peer-reviewed paper 10.3390/ijms242417546