 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dimitar Georgiev Tonev | -- | 5107 | 2023-12-23 18:55:50 | | | |
| 2 | Jason Zhu | Meta information modification | 5107 | 2023-12-25 02:45:55 | | |
Video Upload Options
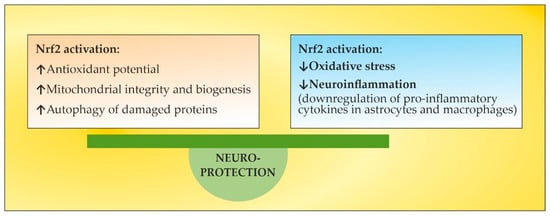
The pathogenesis of multiple sclerosis (MS) suggests that, in genetically susceptible subjects, T lymphocytes undergo activation in the peripheral compartment, pass through the BBB, and cause damage in the CNS. They produce pro-inflammatory cytokines; induce cytotoxic activities in microglia and astrocytes with the accumulation of reactive oxygen species, reactive nitrogen species, and other highly reactive radicals; activate B cells and macrophages and stimulate the complement system. Inflammation and neurodegeneration are involved from the very beginning of the disease. They can both be affected by oxidative stress (OS) with different emphases depending on the time course of MS. Thus, OS initiates and supports inflammatory processes in the active phase, while in the chronic phase it supports neurodegenerative processes. A still unresolved issue in overcoming OS-induced lesions in MS is the insufficient endogenous activation of the Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) pathway, which under normal conditions plays an essential role in mitochondria protection, OS, neuroinflammation, and degeneration. Thus, the search for approaches aiming to elevate endogenous Nrf2 activation is capable of protecting the brain against oxidative damage. However, exogenous Nrf2 activators themselves are not without drawbacks, necessitating the search for new non-pharmacological therapeutic approaches to modulate OS.
1. Introduction
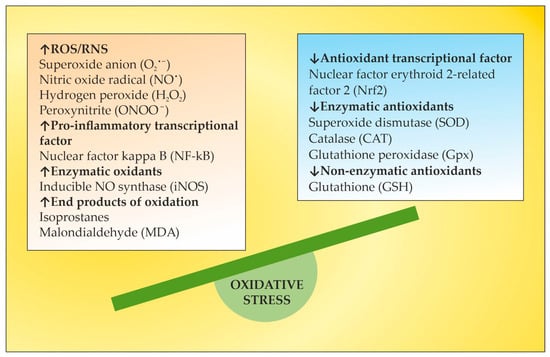
2. OS in MS
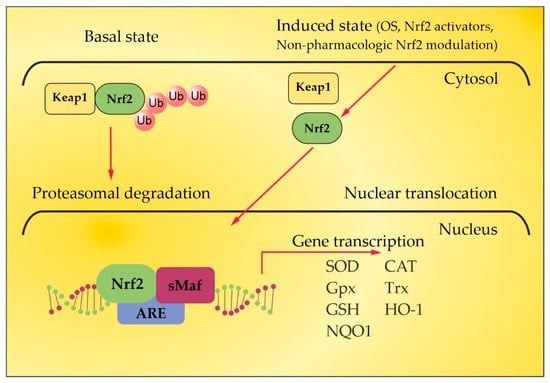
3. Nrf2 Pathway in MS
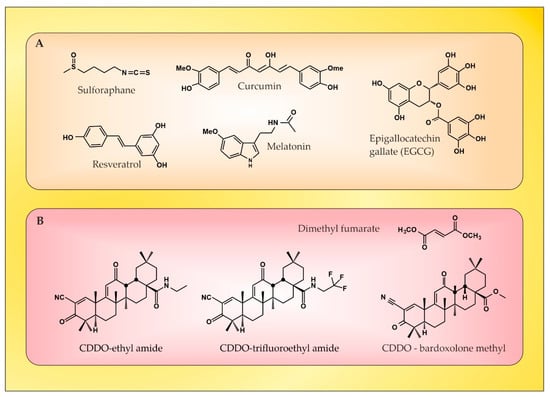
4. The Role of Certain Natural and Synthetic Compounds as Exogenous Nrf2 Activators
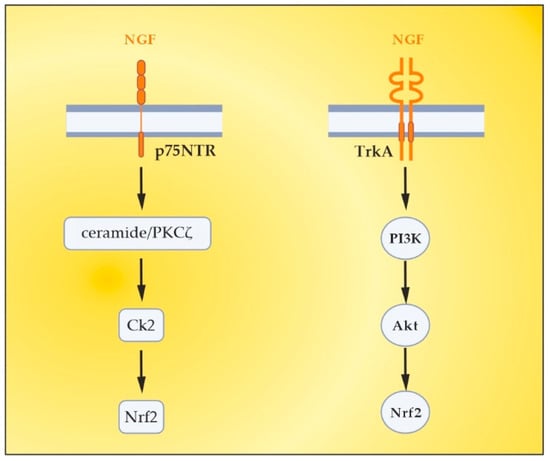
5. The Role of NGF as an Endogenous Nrf2 Activator
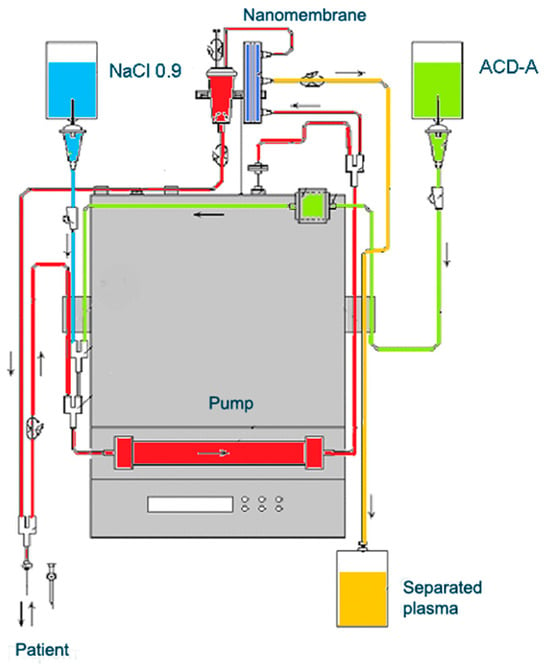
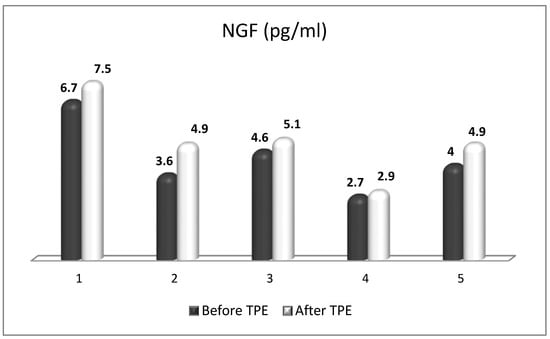
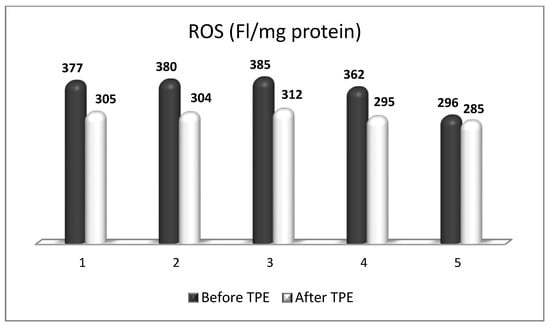
6. The Role of TPE as an OS Modulator
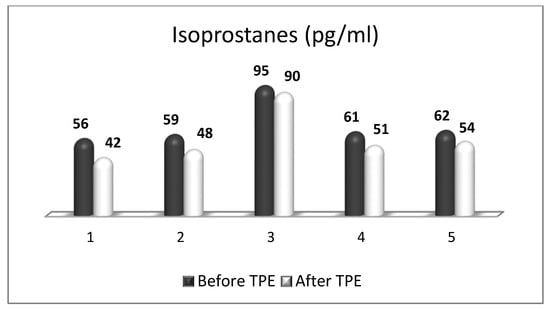
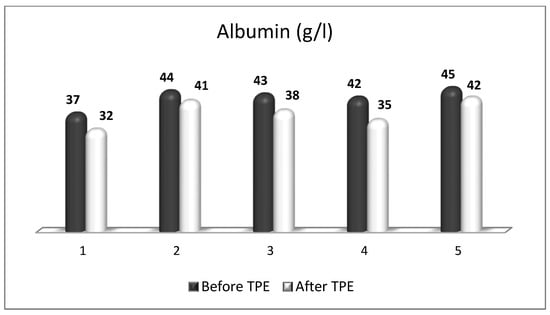
References
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286.
- Lassmann, H.; Brück, W.; Lucchinetti, C.F. The Immunopathology of Multiple Sclerosis: An Overview. Brain Pathol. 2007, 17, 210–218.
- Wang, R.; Liang, L.; Matsumoto, M.; Iwata, K.; Umemura, A.; He, F. Reactive Oxygen Species and NRF2 Signaling, Friends or Foes in Cancer? Biomolecules 2023, 13, 353.
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558.
- Adamczyk, B.; Adamczyk-Sowa, M. New Insights into the Role of Oxidative Stress Mechanisms in the Pathophysiology and Treatment of Multiple Sclerosis. Oxid. Med. Cell Longev. 2016, 2016, 1973834.
- Maldonado, P.P.; Guevara, C.; Olesen, M.A.; Orellana, J.A.; Quintanilla, R.A.; Ortiz, F.C. Neurodegeneration in Multiple Sclerosis: The Role of Nrf2-Dependent Pathways. Antioxidants 2022, 11, 1146.
- Gan, L.; Johnson, J.A. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim. Biophys. Acta 2014, 1842, 1208–1218.
- Amoroso, R.; Maccallini, C.; Bellezza, I. Activators of Nrf2 to Counteract Neurodegenerative Diseases. Antioxidants 2023, 12, 778.
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317.
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590.
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325.
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376.
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, 18.
- Maes, M.; Mihaylova, I.; Kubera, M.; Leunis, J.C.; Geffard, M. IgM-mediated autoimmune responses directed against multiple neoepitopes in depression: New pathways that underpin the inflammatory and neuroprogressive pathophysiology. J. Affect. Disord. 2011, 135, 414–418.
- Maes, M.; Kubera, M.; Mihaylova, I.; Geffard, M.; Galecki, P.; Leunis, J.C.; Berk, M. Increased autoimmune responses against autoepitopes modified by oxidative and nitrosative damage in depression: Implications for the pathways to chronic depression and neuroprogression. J. Affect. Disord. 2013, 149, 23–29.
- Nunes, S.; Vargas, H.; Prado, E.; Barbosa, D.; de Melo, L.; Moylan, S. The shared role of oxidative stress and inflammation in major depressive disorder and nicotine dependence. Neurosci. Biobehav. Rev. 2013, 37, 1336–1345.
- Tonev, D.G.; Momchilova, A.B. Therapeutic Plasma Exchange in Certain Immune-Mediated Neurological Disorders: Focus on a Novel Nanomembrane-Based Technology. Biomedicines 2023, 11, 328.
- Tsonchev, Z.; Alexandrov, A.; Momchilova, A.; Pankov, R.; Orozova, M.; Georgieva, R.; Georgiev, S.; Alexandrov, S.; Voinov, V.; Anaya, F.; et al. Therapeutic Apheresis with Nanotechnology Membrane for Human Diseases; Bulgarian Academy of Science Prof. Marin Drinov Publishing House: Sofia, Bulgaria, 2020; Volume 2, pp. 1–163.
- Ljubisavljevic, S.; Stojanovic, I.; Cvetkovic, T.; Vojinovic, S.; Stojanov, D.; Stefanovic, N.; Pavlovic, D. Erythrocytes’ antioxidative capacity as a potential marker of oxidative stress intensity in neuroinflammation. J. Neurol. Sci. 2014, 337, 8–13.
- Pentón-Rol, G.; Cervantes-Llanos, M.; Martínez-Sánchez, G.; Cabrera-Gómez, J.A.; Valenzuela-Silva, C.M.; Ramírez-Nuñez, O.; Casanova-Orta, M.; Robinson-Agramonte, M.A.; Lopategui-Cabezas, I.; López-Saura, P.A. TNF-α and IL-10 downregulation and marked oxidative stress in Neuromyelitis Optica. J. Inflamm. 2009, 6, 18.
- Jana, A.; Pahan, K. Oxidative Stress Kills Human Primary Oligodendrocytes Via Neutral Sphingomyelinase: Implications for Multiple Sclerosis. J. Neuroimmune Pharmacol. 2007, 2, 184–193.
- Bono, S.; Feligioni, M.; Corbo, M. Impaired antioxidant KEAP1-NRF2 system in amyotrophic lateral sclerosis: NRF2 activation as a potential therapeutic strategy. Mol. Neurodegener. 2021, 16, 71.
- Mahad, D.; Lassmann, H.; Turnbull, D. Review: Mitochondria and disease progression in multiple sclerosis. Neuropathol. Appl. Neurobiol. 2008, 34, 577–589.
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain. 2012, 135, 886–899.
- Haider, L.; Fischer, M.T.; Frischer, J.M.; Bauer, J.; Höftberger, R.; Botond, G.; Esterbauer, H.; Binder, C.J.; Witztum, J.L.; Lassmann, H. Oxidative damage in multiple sclerosis lesions. Brain. 2011, 134, 1914–1924.
- Ortiz, G.G.; Pacheco-Moisés, F.P.; Bitzer-Quintero, O.K.; Ramírez-Anguiano, A.C.; Flores-Alvarado, L.J.; Ramírez-Ramírez, V.; Macias-Islas, M.A.; Torres-Sánchez, E.D. Immunology and Oxidative Stress in Multiple Sclerosis: Clinical and Basic Approach. Clin. Dev. Immunol. 2013, 2013, 708659.
- Amorini, A.M.; Petzold, A.; Tavazzi, B.; Eikelenboom, J.; Keir, G.; Belli, A.; Giovannoni, G.; Di Pietro, V.; Polman, C.; D’Urso, S.; et al. Increase of uric acid and purine compounds in biological fluids of multiple sclerosis patients. Clin. Biochem. 2009, 42, 1001–1006.
- Bjartmar, C.; Trapp, B.D. Axonal and neuronal degeneration in multiple sclerosis: Mechanisms and functional consequences. Curr. Opin. Neurol. 2001, 14, 271–278.
- van der Goes, A.; Wouters, D.; van der Pol, S.M.A.; Huizinga, R.; Ronken, E.; Adamson, P.; Greenwood, J.; Dijkstra, C.D.; de Vries, H.E. Reactive oxygen species enhance the migration of monocytes across the blood-brain barrier in vitro. FASEB J. 2001, 15, 1852–1854.
- Schreibelt, G.; Musters, R.J.P.; Reijerkerk, A.; de Groot, L.R.; van der Pol, S.M.A.; Hendrikx, E.M.L.; Döpp, E.D.; Dijkstra, C.D.; Drukarch, B.; de Vries, H.E. Lipoic Acid Affects Cellular Migration into the Central Nervous System and Stabilizes Blood-Brain Barrier Integrity. J. Immunol. 2006, 177, 2630–2637.
- van der Goes, A.; Brouwer, J.; Hoekstra, K.; Roos, D.; Berg, T.K.V.D.; Dijkstra, C.D. Reactive oxygen species are required for the phagocytosis of myelin by macrophages. J. Neuroimmunol. 1998, 92, 67–75.
- van Meeteren, M.E.; Hendriks, J.; Dijkstra, C.D.; van Tol, E.A. Dietary compounds prevent oxidative damage and nitric oxide production by cells involved in demyelinating disease. Biochem. Pharmacol. 2004, 67, 967–975.
- Hendriks, J.J.; Teunissen, C.E.; de Vries, H.E.; Dijkstra, C.D. Macrophages and neurodegeneration. Brain Res. Rev. 2005, 48, 185–195.
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell Signal. 2007, 19, 1807–1819.
- Gonsette, R.E. Neurodegeneration in multiple sclerosis: The role of oxidative stress and excitotoxicity. J. Neurol. Sci. 2008, 274, 48–53.
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26.
- Voet, S.; Prinz, M.; van Loo, G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol. Med. 2019, 25, 112–123.
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte crosstalk in CNS inflammation. Neuron 2020, 108, 608–622.
- Tobore, T.O. Oxidative/Nitroxidative stress and multiple sclerosis. J. Mol. Neurosci. 2021, 71, 506–514.
- Pegoretti, V.; Swanson, K.A.; Bethea, J.R.; Probert, L.; Eisel, U.; Fischer, R. Inflammation and oxidative stress in multiple sclerosis: Consequences for therapy development. Oxid. Med. Cell. Longev. 2020, 2020, 7191080.
- Varas, R.; Ortiz, F.C. Neuroinflammation in demyelinating diseases: Oxidative stress as a modulator of glial Cross-Talk. Curr. Pharm. Des. 2019, 25, 4755–4762.
- Milatovic, D.; Zaja-Milatovic, S.; Gupta, R.C.; Yu, Y.; Aschner, M. Oxidative damage and neurodegeneration in manganese-induced neurotoxicity. Toxicol. Appl. Pharmacol. 2009, 240, 219–225.
- Halliwell, B.; Chirico, S. Lipid peroxidation: Its mechanism, measurement, and significance. Am. J. Clin. Nutr. 1993, 57, 715S–724S; discussion 724S–725S.
- Halliwell, B. Lipid peroxidation, antioxidants and cardiovascular disease: How should we move forward? Cardiovasc. Res. 2000, 47, 410–418.
- Teunissen, C.E.; Sombekke, M.; van Winsen, L.; Killestein, J.; Barkhof, F.; Polman, C.H.; Dijkstra, C.D.; Blankenstein, M.A.; Pratico, D. Increased plasma 8,12-iso-iPF2alpha- VI levels in relapsing multiple sclerosis patients are not predictive of disease progression. Mult. Scler. 2012, 18, 1092–1098.
- Miller, E.; Mrowicka, M.; Saluk-Juszczak, J.; Ireneusz, M. The level of isoprostanes as a non-invasive marker for in vivo lipid peroxidation in secondary progressive multiple sclerosis. Neurochem. Res. 2011, 36, 1012–1016.
- Ibitoye, R.; Kemp, K.; Rice, C.; Hares, K.; Scolding, N.; Wilkins, A. Oxidative stress-related biomarkers in multiple sclerosis: A review. Biomark. Med. 2016, 10, 889–902.
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65.
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953.
- Katsuoka, F.; Yamamoto, M. Small Maf proteins (MafF, MafG, MafK): History, structure and function. Gene 2016, 586, 197–205.
- Carvalho, A.N.; Firuzi, O.; Gama, M.J.; Horssen, J.V.; Saso, L. Oxidative Stress and Antioxidants in Neurological Diseases: Is There Still Hope? Curr. Drug Targets 2017, 18, 705–718.
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67.
- Cores, A.; Piquero, M.; Villacampa, M.; León, R.; Menéndez, J.C. NRF2 regulation processes as a source of potential drug targets against neurodegenerative diseases. Biomolecules 2020, 10, 904.
- Cores, Á.; Carmona-Zafra, N.; Clerigué, J.; Villacampa, M.; Menéndez, J.C. Quinones as Neuroprotective Agents. Antioxidants 2023, 12, 1464.
- Licht-Mayer, S.; Wimmer, I.; Traffehn, S.; Metz, I.; Brück, W.; Bauer, J.; Bradl, M.; Lassmann, H. Cell type-specific Nrf2 expression in multiple sclerosis lesions. Acta Neuropathol. 2015, 130, 263–277.
- van Horssen, J.; Drexhage, J.A.; Flor, T.; Gerritsen, W.; van der Valk, P.; de Vries, H.E. Nrf2 and DJ1 are consistently upregulated in inflammatory multiple sclerosis lesions. Free Radic. Biol. Med. 2010, 49, 1283–1289.
- Lee, D.H.; Gold, R.; Linker, R.A. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: Therapeutic modulation via fumaric acid esters. Int. J. Mol. Sci. 2012, 13, 11783–11803.
- Lee, J.M.; Li, J.; Johnson, D.A.; Stein, T.D.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Johnson, J.A. Nrf2, a multi-organ protector? FASEB J. 2005, 19, 1061–1066.
- Johnson, D.A.; Amirahmadi, S.; Ward, C.; Fabry, Z.; Johnson, J.A. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol. Sci. 2010, 114, 237–246.
- Larabee, C.M.; Desai, S.; Agasing, A.; Georgescu, C.; Wren, J.D.; Axtell, R.C.; Plafker, S.M. Loss of Nrf2 exacerbates the visual deficits and optic neuritis elicited by experimental autoimmune encephalomyelitis. Mol. Vis. 2016, 22, 1503–1513.
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692.
- Juurlink, B.H.; Thorburne, S.K.; Hertz, L. Peroxide-scavenging deficit underlies oligodendrocyte susceptibility to oxidative stress. Glia. 1998, 22, 371–378.
- Zgorzynska, E.; Dziedzic, B.; Walczewska, A. An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 9592.
- Winyard, P.G.; Blake, D.R. Antioxidants, redox-regulated transcription factors, and inflammation. Adv. Pharmacol. 1997, 38, 403–421.
- Liddell, J.R. Interplay between Nrf2 and NF-κB in Neuroinflammatory Diseases. J. Clin. Cell. Immunol. 2017, 8, 489.
- Michaličková, D.; Hrnčíř, T.; Canová, N.K.; Slanař, O. Targeting Keap1/Nrf2/ARE signaling pathway in multiple sclerosis. Eur. J. Pharmacol. 2020, 873, 172973.
- Schluesener, H.J.; Seid, K. Heme oxygenase-1 in lesions of rat experimental autoimmune encephalomyelitis and neuritis. J. Neuroimmunol. 2000, 110, 114–120.
- Fagone, P.; Patti, F.; Mangano, K.; Mammana, S.; Coco, M.; Touil-Boukoffa, C.; Chikovani, T.; Di Marco, R.; Nicoletti, F. Heme oxygenase-1 expression in peripheral blood mononuclear cells correlates with disease activity in multiple sclerosis. J. Neuroimmunol. 2013, 261, 82–86.
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777.
- Zhang, X.; Guo, J.; Wei, X.; Niu, C.; Jia, M.; Li, Q.; Meng, D. Bach1: Function, Regulation, and Involvement in Disease. Oxid. Med. Cell Longev. 2018, 2018, 1347969.
- Yeh, J.H.; Chen, W.H.; Chiu, H.C.; Bai, C.H. Hemolysis in double-filtration plasmapheresis. Am. J. Clin. Pathol. 2007, 127, 76–80.
- Srinivas, D.; Kamath Sriganesh, K.; Chakrabarti, D.; Venkateswaran, P. Effect of Therapeutic Plasma Exchange on Plasma Constituents in Neurointensive Care Unit Patients: A Retrospective Study. J. Neuroanaesthesiol. Crit. Care 2021, 8, 197–202.
- Ameri, K.; Harris, A.L. Activating transcription factor 4. Int. J. Biochem. Cell Biol. 2008, 40, 14–21.
- Paladino, S.; Conte, A.; Caggiano, R.; Pierantoni, G.M.; Faraonio, R. Nrf2 Pathway in Age-Related Neurological Disorders: Insights into MicroRNAs. Cell Physiol. Biochem. 2018, 47, 1951–1976.
- Gao, S.; Duan, X.; Wang, X.; Dong, D.; Liu, D.; Li, X.; Sun, G.; Li, B. Curcumin attenuates arsenic-induced hepatic injuries and oxidative stress in experimental mice through activation of Nrf2 pathway, promotion of arsenic methylation and urinary excretion. Food Chem. Toxicol. 2013, 59, 739–747.
- Sahin, K.; Orhan, C.; Tuzcu, Z.; Tuzcu, M.; Sahin, N. Curcumin ameloriates heat stress via inhibition of oxidative stress and modulation of Nrf2/HO-1 pathway in quail. Food Chem. Toxicol. 2012, 50, 4035–4041.
- Zeng, C.; Zhong, P.; Zhao, Y.; Kanchana, K.; Zhang, Y.; Khan, Z.A.; Chakrabarti, S.; Wu, L.; Wang, J.; Liang, G. Curcumin protects hearts from FFA-induced injury by activating Nrf2 and inactivating NF-κB both in vitro and in vivo. J. Mol. Cell Cardiol. 2015, 79, 1–12.
- Menon, V.P.; Sudheer, A.R. Antioxidant and anti-inflammatory properties of curcumin. Adv. Exp. Med. Biol. 2007, 595, 105–125.
- Xie, L.; Li, X.K.; Funeshima-Fuji, N.; Kimura, H.; Matsumoto, Y.; Isaka, Y.; Takahara, S. Amelioration of experimental autoimmune encephalomyelitis by curcumin treatment through inhibition of IL-17 production. Int. Immunopharmacol. 2009, 9, 575–581.
- Kanakasabai, S.; Casalini, E.; Walline, C.C.; Mo, C.; Chearwae, W.; Bright, J.J. Differential regulation of CD4(+) T helper cell responses by curcumin in experimental autoimmune encephalomyelitis. J. Nutr. Biochem. 2012, 23, 1498–1507.
- Mohajeri, M.; Sadeghizadeh, M.; Najafi, F.; Javan, M. Polymerized nano-curcumin attenuates neurological symptoms in EAE model of multiple sclerosis through down regulation of inflammatory and oxidative processes and enhancing neuroprotection and myelin repair. Neuropharmacology 2015, 99, 156–167.
- Singh, N.P.; Hegde, V.L.; Hofseth, L.J.; Nagarkatti, M.; Nagarkatti, P. Resveratrol (trans-3,5,4′-trihydroxystilbene) ameliorates experimental allergic encephalomyelitis, primarily via induction of apoptosis in T cells involving activation of aryl hydrocarbon receptor and estrogen receptor. Mol. Pharmacol. 2007, 72, 1508–1521.
- Imler, T.J., Jr.; Petro, T.M. Decreased severity of experimental autoimmune encephalomyelitis during resveratrol administration is associated with increased IL-17+IL-10+ T cells, CD4(−) IFN-gamma+ cells, and decreased macrophage IL-6 expression. Int. Immunopharmacol. 2009, 9, 134–143.
- Gandy, K.A.O.; Zhang, J.; Nagarkatti, P.; Nagarkatti, M. Resveratrol (3,5,4’-Trihydroxy-trans-Stilbene) Attenuates a Mouse Model of Multiple Sclerosis by Altering the miR-124/Sphingosine Kinase 1 Axis in Encephalitogenic T Cells in the Brain. J. Neuroimmune Pharmacol. 2019, 14, 462–477.
- Ghaiad, H.R.; Nooh, M.M.; El-Sawalhi, M.M.; Shaheen, A.A. Resveratrol Promotes Remyelination in Cuprizone Model of Multiple Sclerosis: Biochemical and Histological Study. Mol. Neurobiol. 2017, 54, 3219–3229.
- Sato, F.; Martinez, N.E.; Shahid, M.; Rose, J.W.; Carlson, N.G.; Tsunoda, I. Resveratrol exacerbates both autoimmune and viral models of multiple sclerosis. Am. J. Pathol. 2013, 183, 1390–1396.
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1.
- Yiu, E.M.; Tai, G.; Peverill, R.E.; Lee, K.J.; Croft, K.D.; Mori, T.A.; Scheiber-Mojdehkar, B.; Sturm, B.; Praschberger, M.; Vogel, A.P.; et al. An open-label trial in Friedreich ataxia suggests clinical benefit with high-dose resveratrol, without effect on frataxin levels. J. Neurol. 2015, 262, 1344–1353.
- Aktas, O.; Prozorovski, T.; Smorodchenko, A.; Savaskan, N.E.; Lauster, R.; Kloetzel, P.M.; Infante-Duarte, C.; Brocke, S.; Zipp, F. Green tea epigallocatechin-3-gallate mediates T cellular NF-kappa B inhibition and exerts neuroprotection in autoimmune encephalomyelitis. J. Immunol. 2004, 173, 5794–5800.
- Herges, K.; Millward, J.M.; Hentschel, N.; Infante-Duarte, C.; Aktas, O.; Zipp, F. Neuroprotective effect of combination therapy of glatiramer acetate and epigallocatechin-3-gallate in neuroinflammation. PLoS ONE 2011, 6, e25456.
- Janssen, A.; Fiebiger, S.; Bros, H.; Hertwig, L.; Romero-Suarez, S.; Hamann, I.; Chanvillard, C.; Bellmann-Strobl, J.; Paul, F.; Millward, J.M.; et al. Treatment of Chronic Experimental Autoimmune Encephalomyelitis with Epigallocatechin-3-Gallate and Glatiramer Acetate Alters Expression of Heme-Oxygenase-1. PLoS ONE 2015, 10, e0130251.
- Semnani, M.; Mashayekhi, F.; Azarnia, M.; Salehi, Z. Effects of green tea epigallocatechin-3-gallate on the proteolipid protein and oligodendrocyte transcription factor 1 messenger RNA gene expression in a mouse model of multiple sclerosis. Folia Neuropathol. 2017, 55, 199–205.
- Sun, Q.; Zheng, Y.; Zhang, X.; Hu, X.; Wang, Y.; Zhang, S.; Zhang, D.; Nie, H. Novel immunoregulatory properties of EGCG on reducing inflammation in EAE. Front. Biosci. 2013, 18, 332–342.
- Wang, J.; Pae, M.; Meydani, S.N.; Wu, D. Green tea epigallocatechin-3-gallate modulates differentiation of naïve CD4⁺ T cells into specific lineage effector cells. J. Mol. Med. 2013, 91, 485–495.
- Wu, D. Green tea EGCG, T-cell function, and T-cell-mediated autoimmune encephalomyelitis. J. Investig. Med. 2016, 64, 1213–1219.
- Chesser, A.S.; Ganeshan, V.; Yang, J.; Johnson, G.V. Epigallocatechin-3-gallate enhances clearance of phosphorylated tau in primary neurons. Nutr. Neurosci. 2016, 19, 21–31.
- Bai, Q.; Lyu, Z.; Yang, X.; Pan, Z.; Lou, J.; Dong, T. Epigallocatechin-3-gallate promotes angiogenesis via up-regulation of Nfr2 signaling pathway in a mouse model of ischemic stroke. Behav. Brain. Res. 2017, 321, 79–86.
- Boddupalli, S.; Mein, J.R.; Lakkanna, S.; James, D.R. Induction of phase 2 antioxidant enzymes by broccoli sulforaphane: Perspectives in maintaining the antioxidant activity of vitamins a, C, and e. Front. Genet. 2012, 3, 7.
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and Other Nutrigenomic Nrf2 Activators: Can the Clinician’s Expectation Be Matched by the Reality? Oxid. Med. Cell. Longev. 2016, 2016, 7857186.
- Innamorato, N.G.; Rojo, A.I.; García-Yagüe, A.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008, 181, 680–689.
- Li, B.; Cui, W.; Liu, J.; Li, R.; Liu, Q.; Xie, X.H.; Ge, X.L.; Zhang, J.; Song, X.J.; Wang, Y.; et al. Sulforaphane ameliorates the development of experimental autoimmune encephalomyelitis by antagonizing oxidative stress and Th17-related inflammation in mice. Exp. Neurol. 2013, 250, 239–249.
- Yoo, I.H.; Kim, M.J.; Kim, J.; Sung, J.J.; Park, S.T.; Ahn, S.W. The Anti-Inflammatory effect of sulforaphane in mice with experimental autoimmune encephalomyelitis. J. Korean Med. Sci. 2019, 34, e197.
- Hardeland, R. Aging, Melatonin, and the Pro- and Anti-Inflammatory Networks. Int. J. Mol. Sci. 2019, 20, 1223.
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin: A Versatile Protector against Oxidative DNA Damage. Molecules 2018, 23, 530.
- Deng, Y.; Zhu, J.; Mi, C.; Xu, B.; Jiao, C.; Li, Y.; Xu, D.; Liu, W.; Xu, Z. Melatonin antagonizes Mn-induced oxidative injury through the activation of keap1-Nrf2-ARE signaling pathway in the striatum of mice. Neurotox. Res. 2015, 27, 156–171.
- Ding, K.; Wang, H.; Xu, J.; Li, T.; Zhang, L.; Ding, Y.; Zhu, L.; He, J.; Zhou, M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: The Nrf2-ARE signaling pathway as a potential mechanism. Free Radic. Biol. Med. 2014, 73, 1–11.
- Negi, G.; Kumar, A.; Sharma, S.S. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: Effects on NF-κB and Nrf2 cascades. J. Pineal. Res. 2011, 50, 124–131.
- Long, T.; Yang, Y.; Peng, L.; Li, Z. Neuroprotective Effects of Melatonin on Experimental Allergic Encephalomyelitis Mice Via Anti-Oxidative Stress Activity. J. Mol. Neurosci. 2018, 64, 233–241.
- Miller, E.; Walczak, A.; Majsterek, I.; Kędziora, J. Melatonin reduces oxidative stress in the erythrocytes of multiple sclerosis patients with secondary progressive clinical course. J. Neuroimmunol. 2013, 257, 97–101.
- Pareek, T.K.; Belkadi, A.; Kesavapany, S.; Zaremba, A.; Loh, S.L.; Bai, L.; Cohen, M.L.; Meyer, C.; Liby, K.T.; Miller, R.H.; et al. Triterpenoid modulation of IL-17 and Nrf-2 expression ameliorates neuroinflammation and promotes remyelination in autoimmune encephalomyelitis. Sci. Rep. 2011, 1, 201.
- Wei, H.J.; Pareek, T.K.; Liu, Q.; Letterio, J.J. A unique tolerizing dendritic cell phenotype induced by the synthetic triterpenoid CDDO-DFPA (RTA-408) is protective against EAE. Sci. Rep. 2017, 7, 9886.
- Mrowietz, U.; Morrison, P.J.; Suhrkamp, I.; Kumanova, M.; Clement, B. The Pharmacokinetics of Fumaric Acid Esters Reveal Their In Vivo Effects. Trends Pharmacol. Sci. 2018, 39, 1–12.
- Lin, S.X.; Lisi, L.; Dello Russo, C.; Polak, P.E.; Sharp, A.; Weinberg, G.; Kalinin, S.; Feinstein, D.L. The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro 2011, 3, e00055.
- Gopal, S.; Mikulskis, A.; Gold, R.; Fox, R.J.; Dawson, K.T.; Amaravadi, L. Evidence of activation of the Nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the Phase 3 DEFINE and CONFIRM studies. Mult. Scler. 2017, 23, 1875–1883.
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104.
- Dehmel, T.; Döbert, M.; Pankratz, S.; Leussink, V.I.; Hartung, H.P.; Wiendl, H.; Kieseier, B.C. Monomethylfumarate reduces in vitro migration of mononuclear cells. Neurol. Sci. 2014, 35, 1121–1125.
- Rubant, S.A.; Ludwig, R.J.; Diehl, S.; Hardt, K.; Kaufmann, R.; Pfeilschifter, J.M.; Boehncke, W.H. Dimethylfumarate reduces leukocyte rolling in vivo through modulation of adhesion molecule expression. J. Investig. Dermatol. 2008, 128, 326–331.
- Litjens, N.H.; Rademaker, M.; Ravensbergen, B.; Rea, D.; van der Plas, M.J.; Thio, B.; Walding, A.; van Dissel, J.T.; Nibbering, P.H. Monomethylfumarate affects polarization of monocyte-derived dendritic cells resulting in down-regulated Th1 lymphocyte responses. Eur. J. Immunol. 2004, 34, 565–575.
- Schimrigk, S.; Brune, N.; Hellwig, K.; Lukas, C.; Bellenberg, B.; Rieks, M.; Hoffmann, V.; Pöhlau, D.; Przuntek, H. Oral fumaric acid esters for the treatment of active multiple sclerosis: An open-label, baseline-controlled pilot study. Eur. J. Neurol. 2006, 13, 604–610.
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097.
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107.
- Xu, Z.; Zhang, F.; Sun, F.; Gu, K.; Dong, S.; He, D. Dimethyl fumarate for multiple sclerosis. Cochrane Database Syst. Rev. 2015, 4, CD011076.
- Ermis, U.; Weis, J.; Schulz, J.B. PML in a patient treated with fumaric acid. N. Engl. J. Med. 2013, 368, 1657–1658.
- Nieuwkamp, D.J.; Murk, J.L.; van Oosten, B.W.; Cremers, C.H.; Killestein, J.; Viveen, M.C.; Van Hecke, W.; Frijlink, D.W.; Wattjes, M.P.; PML in Dutch MS Patients Consortium. PML in a patient without severe lymphocytopenia receiving dimethyl fumarate. N. Engl. J. Med. 2015, 372, 1474–1476.
- Rosenkranz, T.; Novas, M.; Terborg, C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N. Engl. J. Med. 2015, 372, 1476–1478.
- Hammer, A.; Waschbisch, A.; Kuhbandner, K.; Bayas, A.; Lee, D.H.; Duscha, A.; Haghikia, A.; Gold., R.; Linker, R.A. The NRF2 pathway as potential biomarker for dimethyl fumarate treatment in multiple sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 668–676.
- Martín-Montañez, E.; Pavia, J.; Valverde, N.; Boraldi, F.; Lara, E.; Oliver, B.; Hurtado-Guerrero, I.; Fernandez, O.; Garcia-Fernandez, M. The S1P mimetic fingolimod phosphate regulates mitochondrial oxidative stress in neuronal cells. Free Radic. Biol. Med. 2019, 137, 116–130.
- Colombo, E.; Bassani, C.; De Angelis, A.; Ruffini, F.; Ottoboni, L.; Comi, G.; Martino, G.; Farina, C. Siponimod (BAF312) Activates Nrf2 While Hampering NFκB in Human Astrocytes, and Protects from Astrocyte-Induced Neurodegeneration. Front. Immunol. 2020, 11, 635.
- Tasset, I.; Bahamonde, C.; Agüera, E.; Conde, C.; Cruz, A.H.; Pérez-Herrera, A.; Gascón, F.; Giraldo, A.I.; Ruiz, M.C.; Lillo, R.; et al. Effect of natalizumab on oxidative damage biomarkers in relapsing-remitting multiple sclerosis. Pharmacol. Rep. 2013, 65, 624–631.
- Levi-Montalcini, R.; Skaper, S.D.; Dal Toso, R.; Petrelli, L.; Leon, A. Nerve growth factor: From neurotrophin to neurokine. Trends Neurosci. 1996, 19, 514–520.
- Barbacid, M. Neurotrophic factors and their receptors. Curr. Opin. Cell Biol. 1995, 7, 148–155.
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309.
- Chao, M.V.; Hempstead, B.L. p75 and Trk: A two-receptor system. Trends Neurosci. 1995, 18, 321–326.
- Lorenzini, L.; Baldassarro, V.A.; Stanzani, A.; Giardino, L. Nerve Growth Factor: The First Molecule of the Neurotrophin Family. Adv. Exp. Med. Biol. 2021, 1331, 3–10.
- Loy, R.; Taglialatela, G.; Angelucci, L.; Heyer, D.; Perez-Polo, R. Regional CNS uptake of blood-borne nerve growth factor. J. Neurosci. Res. 1994, 39, 339–346.
- Tiberi, A.; Carucci, N.M.; Testa, G.; Rizzi, C.; Pacifico, P.; Borgonovo, G.; Arisi, I.; D’onofrio, M.; Brandi, R.; Gan, W.-B.; et al. Reduced levels of NGF shift astrocytes toward a neurotoxic phenotype. Front. Cell Dev. Biol. 2023, 11, 1165125.
- Rizzi, C.; Tiberi, A.; Giustizieri, M.; Marrone, M.C.; Gobbo, F.; Carucci, N.M.; Meli, G.; Arisi, I.; D’Onofrio, M.; Marinelli, S.; et al. NGF steers microglia toward a neuroprotective phenotype. Glia 2018, 66, 1395–1416.
- Villoslada, P.; Genain, C.P. Role of nerve growth factor and other trophic factors in brain inflammation. Prog. Brain Res. 2004, 146, 403–414.
- Guarnieri, G.; Sarchielli, E.; Comeglio, P.; Herrera-Puerta, E.; Piaceri, I.; Nacmias, B.; Benelli, M.; Kelsey, G.; Maggi, M.; Gallina, P.; et al. Tumor necrosis factor α influences phenotypic plasticity and promotes epigenetic changes in human basal forebrain cholinergic neuroblasts. Int. J. Mol. Sci. 2020, 21, 6128.
- Micera, A.; Properzi, F.; Triaca, V.; Aloe, L. Nerve growth factor antibody exacerbates neuropathological signs of experimental allergic encephalomyelitis in adult Lewis rats. J. Neuroimmunol. 2000, 104, 116–123.
- Villoslada, P.; Hauser, S.L.; Bartke, I.; Unger, J.; Heald, N.; Rosenberg, D.; Cheung, S.W.; Mobley, W.C.; Fisher, S.; Genain, C.P. Human nerve growth factor protects common marmosets against autoimmune encephalomyelitis by switching the balance of t helper cell type 1 and 2 cytokines within the central nervous system. J. Exp. Med. 2000, 191, 1799–1806.
- Kenarov, P.; Petrov, N.; Voinov, V.; Daskalov, M.; Anaya, F.; Russo, G.; Momchilova, A. A new approach using nanomem-brane—Based therapeutic plasmapheresis for treatment of patients with multiple sclerosis. A case report. J. Pharmacol. Clin. Toxicol. 2014, 2, 1031.
- Tonev, D.; Momchilova, A. Therapeutic Plasma Exchange and Multiple Sclerosis Dysregulations: Focus on the Removal of Pathogenic Circulatory Factors and Altering Nerve Growth Factor and Sphingosine-1-Phosphate Plasma Levels. Curr. Issues Mol. Biol. 2023, 45, 7749–7774.
- Satoh, T.; Sakai, N.; Enokido, Y.; Uchiyama, Y.; Hatanaka, H. Free radical-independent protection by nerve growth factor and Bcl-2 of PC12 cells from hydrogen peroxide-triggered apoptosis. J. Biochem. 1996, 120, 540–546.
- Dugan, L.L.; Creedon, D.J.; Johnson, E.M., Jr.; Holtzman, D.M. Rapid suppression of free radical formation by nerve growth factor involves the mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. USA 1997, 94, 4086–4091.
- Tang, L.-L.; Wang, R.; Tang, X.-C. Huperzine A Protects SHSY5Y Neuroblastoma Cells against Oxidative Stress Damage via Nerve Growth Factor Production. Eur. J. Pharmacol. 2005, 519, 9–15.
- Sampath, D.; Perez-Polo, R. Regulation of antioxidant enzyme expression by NGF. Neurochem. Res. 1997, 22, 351–362.
- Salinas, M.; Diaz, R.; Abraham, N.G.; Ruiz de Galarreta, C.M.; Cuadrado, A. Nerve growth factor protects against 6-hydroxydopamine-induced oxidative stress by increasing expression of heme oxygenase-1 in a phosphatidylinositol 3-kinase-dependent manner. J. Biol. Chem. 2003, 278, 13898–13904.
- Rojo, A.I.; Salinas, M.; Martín, D.; Perona, R.; Cuadrado, A. Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-kappaB. J. Neurosci. 2004, 24, 7324–7334.
- Mimura, J.; Kosaka, K.; Maruyama, A.; Satoh, T.; Harada, N.; Yoshida, H.; Satoh, K.; Yamamoto, M.; Itoh, K. Nrf2 regulates NGF mRNA induction by carnosic acid in T98G glioblastoma cells and normal human astrocytes. J. Biochem. 2011, 150, 209–217.
- Kosaka, K.; Mimura, J.; Itoh, K.; Satoh, T.; Shimojo, Y.; Kitajima, C.; Maruyama, A.; Yamamoto, M.; Shirasawa, T. Role of Nrf2 and p62/ZIP in the neurite outgrowth by carnosic acid in PC12h cells. J. Biochem. 2010, 147, 73–81.
- Su, R.; Su, W.; Jiao, Q. NGF protects neuroblastoma cells against β-amyloid-induced apoptosis via the Nrf2/HO-1 pathway. FEBS Open Bio 2019, 9, 2063–2071, Erratum in: FEBS Open Bio 2020, 10, 288.
- Ishii, T.; Warabi, E.; Mann, G.E. Circadian control of BDNF-mediated Nrf2 activation in astrocytes protects dopaminergic neurons from ferroptosis. Free Radic. Biol. Med. 2019, 133, 169–178.
- Valdovinos-Flores, C.; Limón-Pacheco, J.H.; León-Rodríguez, R.; Petrosyan, P.; Garza-Lombó, C.; Gonsebatt, M.E. Systemic L-Buthionine -S-R-Sulfoximine Treatment Increases Plasma NGF and Upregulates L-cys/L-cys2 Transporter and γ-Glutamylcysteine Ligase mRNAs Through the NGF/TrkA/Akt/Nrf2 Pathway in the Striatum. Front. Cell Neurosci. 2019, 13, 325.
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523.
- Levi-Montalcini, R.; Aloe, L. Differentiating effects of murine nerve growth factor in the peripheral and central nervous systems of Xenopus laevis tadpoles. Proc. Natl. Acad. Sci. USA 1985, 82, 7111–7115.
- Connelly-Smith, L.; Alquist, C.R.; Aqui, N.A.; Hofmann, J.C.; Klingel, R.; Onwuemene, O.A.; Patriquin, C.J.; Pham, H.P.; Sanchez, A.P.; Schneiderman, J.; et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice—Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Ninth Special Issue. J. Clin. Apher. 2023, 38, 77–278.
- Redant, S.; De Bels, D.; Ismaili, K.; Honoré, P.M. Membrane-based therapeutic plasma exchange in intensive care. Blood Purif. 2021, 50, 290–297.
- Reeves, H.M.; Winters, J.L. The mechanisms of action of plasma exchange. Br. J. Haematol. 2014, 164, 342–351.
- Yamakova, Y.; Ilieva, V.A.; Petkov, R.; Yankov, G. Nanomembrane-Based Therapeutic Plasmapheresis after Non-Invasive Ventilation Failure for Treatment of a Patient with Acute Respiratory Distress Syndrome and Myasthenia Gravis: A Case Re-port. Blood Purif. 2019, 48, 382–384.
- Alexandrov, A.; Vassileva, P.; Momchilova, A.; Tsonchev, Z.; Kirilova, Y.; Ivanova, R.; Sapundzhiev, P.; Petkova, D.; Tzoneva, R.; Daskalov, M.; et al. A new approach using nanomembrane-based therapeutic plasmapheresis for treatment of patients with multiple sclerosis and neuromyelitis optica. Comptes Rendus L’academie Bulg. Sci. 2016, 69, 373–384.
- Momchilova, A.; Tsonchev, Z.; Hadzhilazova, M.; Tzoneva, R.; Alexandrov, A.; Nikolakov, D.; Ilieva, V.; Pankov, R. Sphin-golipid Metabolism Is Dysregulated in Erythrocytes from Multiple Sclerosis Patient. Comptes Rendus L’academie Bulg. Sci. 2020, 73, 426–432.
- Sapundzhiev, P.; Momchilova, A.; Vassileva, P.; Kirilova, Y.; Ivanova, R.; Bozhilova, M.; Orozova, M.; Staneva, G.; Krastev, P.; Pankov, R.; et al. Plasmapheresis Affects Ophthalmological Parameters and Oxidative Stress in Patients with Multiple Sclerosis and Neuromyelitis Optica. Arch. Biomed. Eng. Biotechnol. 2021, 5, 000617.
- Tenchov, B.; Koynova, R.; Antonova, B.; Zaharinova, S.; Abarova, S.; Tsonchev, Z.; Komsa-Penkova, R.; Momchilova, A. Blood plasma thermal behavior and protein oxidation as indicators of multiple sclerosis clinical status and plasma exchange therapy progression. Thermochim. Acta 2019, 671, 193–199.
- Tonev, D.; Georgieva, R.; Vavrek, E. Our Clinical Experience in the Treatment of Myasthenia Gravis Acute Exacerbations with a Novel Nanomembrane-Based Therapeutic Plasma Exchange Technology. J. Clin. Med. 2022, 11, 4021.
- Escolar, G.; Páez, A.; Cid, J. Conventional Therapeutic Plasma Exchange Versus Low Volume Plasma Exchange in Chronic Pathologies: Potential Benefit in Alzheimer’s Disease. Plasmatology 2022, 16, 26348535221131685.
- Klingele, M.; Allmendinger, C.; Thieme, S.; Baerens, L.; Fliser, D.; Jan, B. Therapeutic apheresis within immune-mediated neurological disorders: Dosing and its effectiveness. Sci. Rep. 2020, 10, 7925.
- Dorst, J.; Fillies, F.; Dreyhaupt, J.; Senel, M.; Tumani, H. Safety and Tolerability of Plasma Exchange and Immunoadsorption in Neuroinflammatory Diseases. J. Clin. Med. 2020, 9, 2874.
- Padmanabhan, A.; Connelly-Smith, L.; Aqui, N.; Balogun, R.A.; Klingel, R.; Meyer, E.; Pham, H.P.; Schneiderman, J.; Witt, V.; Wu, Y.; et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice—Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Eighth Special Issue. J. Clin. Apher. 2019, 34, 171–354.
- Zhang, S.Y.; Gui, L.N.; Liu, Y.Y.; Shi, S.; Cheng, Y. Oxidative Stress Marker Aberrations in Multiple Sclerosis: A Meta-Analysis Study. Front. Neurosci. 2020, 14, 823.
- LeVine, S.M. Albumin and multiple sclerosis. BMC Neurol. 2016, 16, 47.
- Xue, H.; Yang, Z.; Wang, L.; Jiang, Y.; Li, J.; Wu, M.; Wang, G.; Zhang, Y.; Zhang, M. Factors Influencing the Degree of Disability in Patients with Multiple Sclerosis. Front. Neurol. 2021, 12, 714631.
- Boss, K.; Stettner, M.; Szepanowski, F.; Mausberg, A.K.; Paar, M.; Pul, R.; Kleinschnitz, C.; Oettl, K.; Kribben, A. Severe and long-lasting alteration of albumin redox state by plasmapheresis. Sci. Rep. 2022, 12, 12165.
- Alexandrov, A.; Momchilova, A.; Orozova, M.; Alexandrov, S.; Krastev, P.; Stanev, G.; Nikolova, B.; Tsonchev, Z. Therapeutic Apheresis; Bulgarian Academy of Science Prof. Marin Drinov Publishing House: Sofia, Bulgaria, 2022; pp. 1–120.


