The pathogenesis of multiple sclerosis (MS) suggests that, in genetically susceptible subjects, T lymphocytes undergo activation in the peripheral compartment, pass through the BBB, and cause damage in the CNS. They produce pro-inflammatory cytokines; induce cytotoxic activities in microglia and astrocytes with the accumulation of reactive oxygen species, reactive nitrogen species, and other highly reactive radicals; activate B cells and macrophages and stimulate the complement system. Inflammation and neurodegeneration are involved from the very beginning of the disease. They can both be affected by oxidative stress (OS) with different emphases depending on the time course of MS. Thus, OS initiates and supports inflammatory processes in the active phase, while in the chronic phase it supports neurodegenerative processes. A still unresolved issue in overcoming OS-induced lesions in MS is the insufficient endogenous activation of the Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) pathway, which under normal conditions plays an essential role in mitochondria protection, OS, neuroinflammation, and degeneration. Thus, the search for approaches aiming to elevate endogenous Nrf2 activation is capable of protecting the brain against oxidative damage. However, exogenous Nrf2 activators themselves are not without drawbacks, necessitating the search for new non-pharmacological therapeutic approaches to modulate OS.
- multiple sclerosis
- oxidative stress modulation
- Nuclear Factor Erythroid 2-Related Factor 2 pathway activation
1. Introduction
23. OS in MS
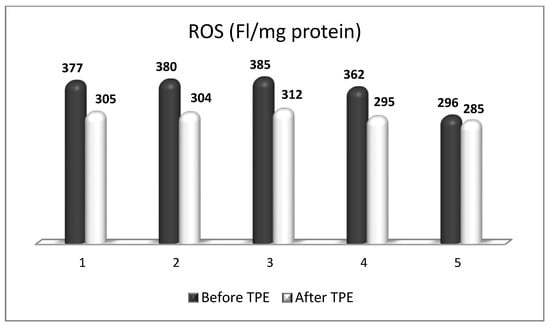
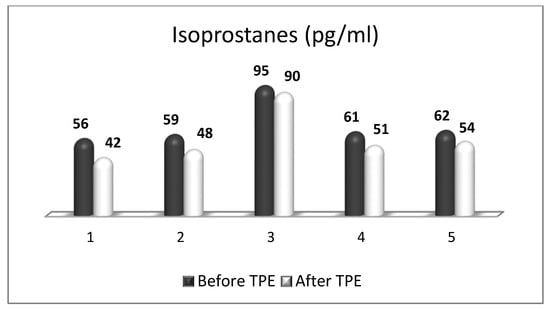
OS plays a key role in neurodegenerative processes by inducing oxidative damage to lipid, protein, and deoxyribonucleic acid (DNA) molecules. Oxidative destruction of proteins makes them alter their active configuration and form oligomers with different functions or molecular fragments that cause pro-inflammatory processes that aggravate OS. Mitochondrial DNA also undergoes alterations caused by ROS and RNS, leading to misfunctions in the key proteins involved in key cellular metabolic processes. The impairment of mitochondrial activity leads to the accumulation of ROS and activation of pro-apoptotic pathways. Lipid peroxidation caused by ROS destroys the structure, asymmetry, and permeability of cell membranes’ bilayer, leading to an enhanced inflammatory response, changes in calcium homeostasis, and neuronal death [11][12][13][11,12,13]. Moreover, the degree of oxidative attack towards proteins, lipids, and DNA frequently is sufficient to generate neoepitopes due to a loss of immunological tolerance, leading to the development of autoimmunity [14][15][16][14,15,16]. The latter implication increases the relevance of using TPE as a means of both modulating OS and ameliorating its adverse consequences. OS is a well-recognized pathogenic factor in the onset and development of a vast variety of neurodegenerative pathologies, including MS. This neurodegenerative disease with a pronounced neuroinflammatory component is characterized by different symptoms and clinical manifestations, which are basically accompanied by inflammation-related damage to the CNS and different levels of disturbed motor activity in affected patients [17]. Inflammation elevates the content of free radicals, causing a higher number of oxidative stress biomarkers [18]. OS is generally an impaired oxidant/antioxidant balance that can cause neuroinflammation and neurodegeneration [19]. The imbalance is characterized by increases in ROS/RNS, the pro-inflammatory transcriptional factor, enzymatic oxidants, and oxidation end products at the expense of decreases in the antioxidant transcriptional factor and enzymatic and non-enzymatic antioxidants (Figure 1). Since OS may be associated with neurodegenerative disorders, an antioxidant-related therapeutic approach might be a promising perspective [20]. OS has also been assumed to be a significant pathogenic factor underlying the etiology of MS [21].
34. Nrf2 Pathway in MS
Under homeostatic conditions, Nrf2 is localized in the cytosol through its negative regulator, kelch-like ECH-associated protein 1 (Keap1), and is subject to proteasomes disintegration through a couple of ubiquitin ligase systems [48][49][48,49]. Thus, the Keap1/Nrf2 interaction underlies a low expression of Nrf2-regulated genes. OS or Nrf2 activators, along with non-pharmacologic Nrf2 modulation, break the complex between Nrf2 and Keap1, inducing the translocation of Nrf2 towards the nucleus. There, Nrf2 heterodimerizes with small musculoaponeurotic fibrosarcoma (sMaf) proteins and binds to antioxidant response elements (ARE) in the promoter region of target genes [48][49][50][51][52][48,49,50,51,52]. This region encodes several antioxidant enzymes that neutralize ROS and electrophiles, involving superoxide dismutase (SOD), glutathione peroxidase (GPx), catalase, glutathione reductase, and heme oxygenase-1 (HO-1) (Figure 2) [53][54][53,54]. Autopsy specimens of MS patients show the upregulation of Nrf2 and the Nrf2-responsive genes HO-1 and NQO-1, in and near to the active lesions in spinal cord and brain samples [55][56][55,56], as an integral part of the cellular anti-oxidative response. The increased expression of Nrf2 has been observed in astrocytes and macrophages in active lesions [54] and in oligodendrocytes at the lesion’s edges [55]. However, the level of Nrf2 expression is cell-type-specific [57]. In the CNS, astrocytes harbor a more efficient anti-oxidative potential compared to neurons. The expression of Nrf2 is lower in neurons, even when they are surrounded by Nrf2-positive glia [56]. Consistent with the essential role of Nrf2 for ROS detoxification, Nrf2-deficient cells are more susceptible to oxidative destruction, etc. [58]. This may underlie a limited capacity of neurons to cope with OS. In experimental autoimmune encephalomyelitis (EAE), an established model for MS, Nrf2-deficient mice show a more rapid onset, an exacerbated clinical severity, an increased number of lesions and infiltrating immune cells, greater microglial activation, and visual dysfunction [59][60][61][59,60,61]. Additionally, and importantly, the oligodendrocytes damaged in the EAE lesions have relatively low levels of Nrf2. Therefore, low levels of Nrf2 or the impaired activation of Nrf2 in oligodendrocytes may account for selective susceptibility in neuroinflammatory conditions due to the high vulnerability of these oligodendrocytes to OS [62]. Dysfunctions in the Nrf2 signaling pathway result in the impairment of redox homeostasis, which leads to ROS/RNS overload [63] and the prevalence of other redox-sensitive transcription factors such as activator protein 1 and the pro-inflammatory nuclear factor kappa-light chain-enhancer of activated B cells (NF-kB). The latter stimulates the expression of certain genes involved in MS pathogenesis, such as tumor necrosis factor α (TNF-α), iNOS, interleukin 1α/β (IL-1α/β), and some growth factors [64]. There is a crosstalk between the Nrf2 and NF-ĸB pathways. Nrf2 binds with its transcriptional cofactor cAMP-response-element-binding protein (CBP) to initiate ARE-driven gene expression. When NF-ĸB binds with CBP in a competitive way, it hinders the binding between CBP and Nrf2, the latter leading to inhibition of Nrf2 activation. Thus, NF-kB and Nrf2 compete for CBP, which promotes DNA binding [65]. Given that OS and inflammation are closely related processes in MS, the activation of the Nrf2 pathway interferes with both processes in a complex manner [66]. HO-1, one of the most important antioxidant enzymes downstream of Nrf2, plays an important role for the anti-inflammatory activity in EAE and MS [67]. Decreased expression of HO-1 was found in peripheral blood mononuclear cells (PBMCs) of MS patients, and a significant downregulation of this enzyme was observed during disease exacerbations [68]. These findings indicate a higher likelihood of relapse in patients with reduced HO-1 expression in PBMC and are confirmed by the results of a microarray meta-analysis [68]. In this context, HO-1 inducers may occur as important factors in the treatment of MS. A thorough understanding of Nrf2-mediated HO-1 gene transactivation requires taking into account the cooperation or competition with other transcriptional factors at ARE and ARE-like sites [69]. The activating transcription factor 4 (ATF4) can dimerize with Nrf2 at ARE to promote the expression of HO-1, whereas the transcription repressor BTB and CNC homology 1 (BACH1), which are critical in the regulation of the HO-1 gene, compete with Nrf2 at the ARE binding sites, and Nrf2-induced expression requires the inactivation of BACH1 sites [69]. In addition, the expression of HO-1 is downregulated by BACH1 when the heme content is low, but higher heme levels inhibit BACH1–DNA binding and promote BACH1 exportation and degradation [70]. TPE techniques using peripheral venous access and high transmembrane pressure cause hemolysis during the procedure [71], which could contribute to BACH1 inhibition as well. Likewise, the significant decrease in plasma proteins and other plasma constituents observed during TPE [72] could contribute to amino acid losses, a well-known inducer of ATF4 [73]. Thus, TPE could modulate HO-1 expression indirectly at multiple levels. In summary, OS and antioxidant Nrf2 pathways are important players in the pathophysiology of MS and represent a promising target for approved or investigational pharmacological and non-pharmacological therapies of MS [57].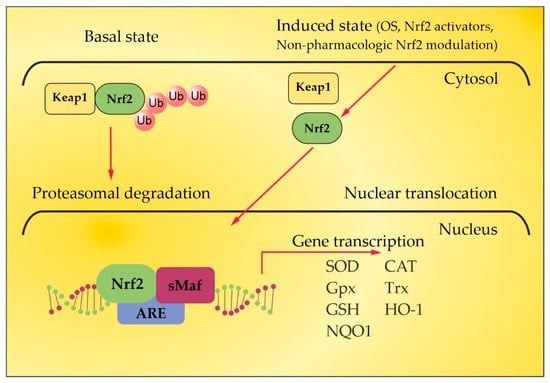
45. The Role of Certain Natural and Synthetic Compounds as Exogenous Nrf2 Activators
The redox homeostasis in the brain tissue is under the control of Nrf2, and there has been a growing interest in identifying natural or synthetic compounds (Figure 3) that are able to modify Nrf2 activity, for example, in MS, in which ROS/RNS have a recognized function [74].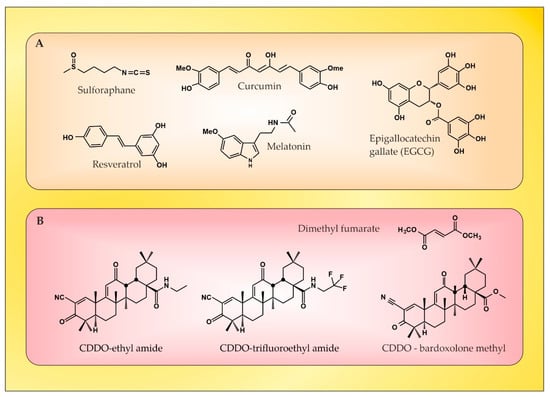
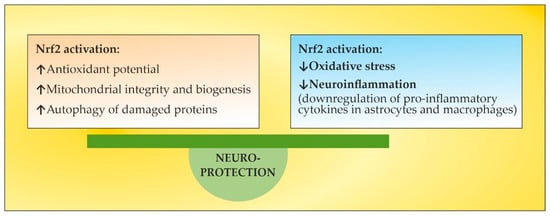
56. The Role of NGF as an Endogenous Nrf2 Activator
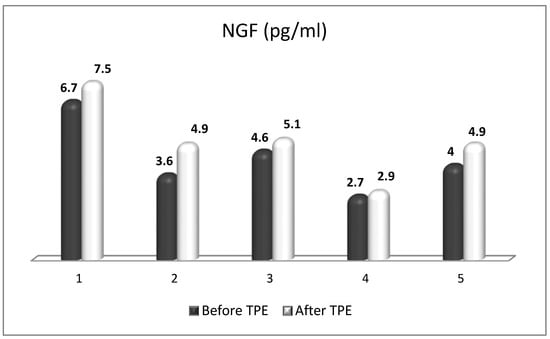
NGF is the most recognized neurotrophin, and it was described in 1952 by Levi-Montalcini [130]. NGF activates two types of membrane receptors, tropomyosin receptor kinase A (TrkA) and p75 neurotrophin receptor (p75NTR) [131]. Depending on the NGF-specific receptors, signaling pathways associated with neuronal differentiation, maturation and survival, axonal and dendrite development, or apoptosis can be stimulated. When TrkA and p75NTR are co-expressed, they comprise a two-receptor heterotetrameric complex that binds to NGF, resulting in the activation of various signaling pathways [132][133][132,133]. In neurological disorders with neuroinflammation, almost all resident cells of the CNS overexpress NGF [134]. Moreover, NGF can cross the BBB when the BBB becomes permeable in pathological conditions, such as MS [135]. It is noteworthy that NGF levels influence glial physiology. There is evidence that in an in vivo mouse model [136], the depletion of NGF causes pro-inflammatory astrocyte activation and neurotoxicity. Conversely, the upregulation of NGF directs microglia to an anti-inflammatory phenotype [137], thereby leading to neuroprotection. The latter is of particular importance in the context of OS in MS, considering the role of pro-inflammatory microglia and astrocytes in the generation of ROS/RNS. Previous studies have observed the significant effects of the intracranial administration of NGF on the downregulation of cytokines (important factors in OS) that are specific to the CNS parenchyma and not detected in the periphery [138]. In the case of MS, during acute attacks, patients show increased levels of NGF in CSF compared to healthy individuals, which can be seen as an attempt to protect the CNS tissue against inflammation [139]. There, observations imply the relevance of NGF-related therapeutic schemes in patients with inflammatory and neurodegenerative pathologies [136]. Last but not least, NGF antibodies have been observed to exacerbate the neuropathological signs of EAE [140]. This suggests not only the importance of NGF in decreasing the EAE lesions [141], but also offers new possibilities for increasing the anti-inflammatory potential of NGF in MS patients by removing these antibodies using TPE [142][143][142,143]. Besides the already-mentioned anti-inflammatory potential, NGF plays an important role in antioxidant protection in experimental and clinical settings. As early as the 1990s, preclinical studies outlined the role of NGF in reducing OS [144][145][144,145]. The activation of the MAPK pathway appears to be required for this action of NGF (namely acute suppression of neuronal ROS production by NGF), implicating TrkA signaling in defense against oxidative damage [145][146][145,146]. Other research from the late 1990s and the early next decade revealed the role of NGF in the upregulation of enzymatic antioxidants (SOD, HO-1, GPx, and catalase) through phosphoinositide-3-kinase (PI3K)/Akt and NF-kB signaling [22][147][148][149][22,147,148,149]. More recent studies have shown that the activation of TrkA by NGF induces the nuclear translocation of Nrf2 and subsequently induces the transcription of ARE-containing genes [150][151][152][150,151,152]. Moreover, the activated Nrf2 can in turn up-regulate NGF gene expression and a positive feedback loop between Nrf2 and NGF can be formed [150][151][150,151], which further strengthens the evidence for the potential of NGF as an Nrf2-inducer. New evidence suggests that NGF is able to activate p75NTR/ceramide-protein kinase C-ζ (PKCζ)/casein kinase 2 (CK2) signaling pathways through direct interaction with p75NTR in order to mediate its activation of Nrf2 [153] (Figure 5). Most interesting in view of future clinical benefits is a recent preclinical study whose results provide evidence that the depletion of peripheral GSH pools increases peripheral circulating NGF, which orchestrates a neuroprotective response in the CNS, at least in the striatum, via the NGF/TrkA/Akt/Nrf2 pathway [154]. A plausible explanation for the role of NGF after systemic OS suggests an increased synthesis of NGF in peripheral tissues, followed by secretion and increased levels in the systemic circulation. Peripheral NGF then reaches the cerebral circulation and activates the TrkA pathway in brain endothelial cells, which induces the transcription of sulfhydryl AAs L-cys/L-cys2 transporters in neurons and glial cells. Given that brain GSH synthesis is limited by the presence of the sulfhydryl AAs L-cys and L-cys2 [155], it has been suggested that the activation of the NGF/TrkA/Akt/Nrf2 signaling pathway in the striatum may be related to the transcriptional regulation of amino acid transporter genes associated with the presence of L-cys and L-cys2 in the brain. In addition, NGF synthesis is elevated in brain endothelial cells, and NGF is secreted into the brain parenchyma, activating the NGF/TrkA/PI3K/Akt/Nrf2 pathway, which stimulates the expression of antioxidant genes in the CNS. Another possibility is that peripheral NGF enters the CNS directly, as suggested in the 1980s by Levi-Montalcini [156], which may occur with a permeable BBB, as is the case in acute exacerbations of MS [135][143][135,143]. Translating the results of this preclinical model to the real clinical setting is achievable through the application of TPE in the treatment of acute exacerbations of MS. Through this non-pharmacological therapeutic approach, both an increase in circulating levels of NGF and a modulation of OS can be achieved [17][143][17,143]. Evidence for this will be presented in the next section.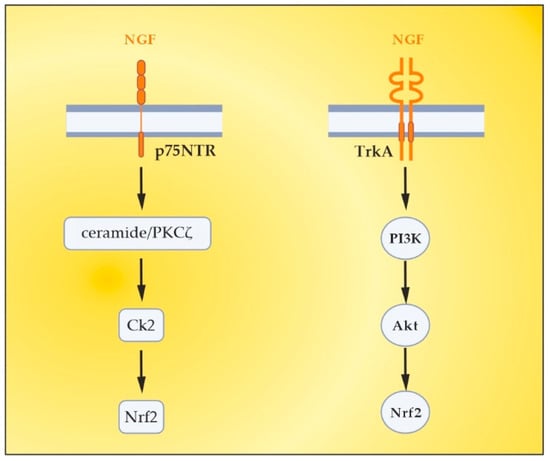
67. The Role of TPE as an OS Modulator
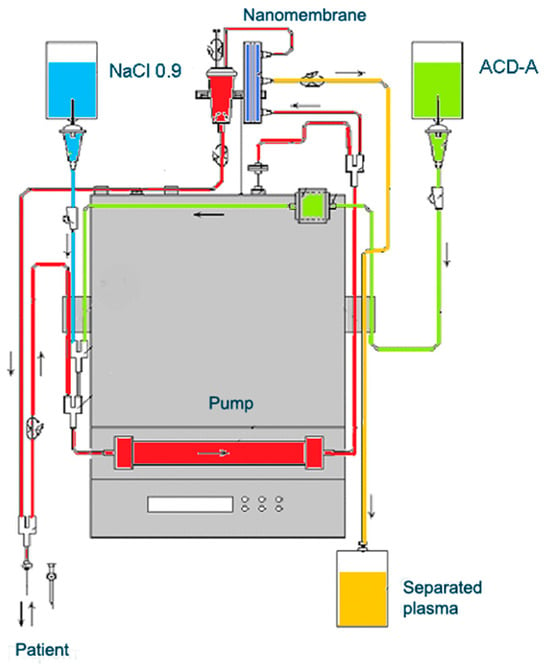
TPE is a therapeutic approach that involves extracorporeal blood removal, as well as an exchange of blood plasma or components. It basically removes a significant volume of plasma (1 to 1.5 of the patient’s total plasma volume (TPV) per treatment) which is accompanied with corresponding volume replacement using colloid solutions or a combination of crystalloid/colloid solutions [157]. The aim of TPE is to remove potentially pathogenic substances with high molecular weight (>150 kDA), involving autoantibodies, pro-inflammatory mediators, lipids, and other molecules or molecular fragments from the intravascular space [157][158][157,158]. However, the mechanism of TPE action in inflammatory and neurodegenerative disorders involves more than just a removal of potentially a number of pathogenic molecules. It should be noted that TPE may also modulate cellular immunological response by changing the ratio between T helper type-1 (Th-1) and type-2 (Th-2) cells. Th-2 cells regulate the humoral immune response by facilitating B-cell autoantibody production and play an important role in the development of neurodegenerative autoimmune pathologies. By shifting the balance between peripheral T cells from Th-2 to Th-1 predominance, TPE modulates the pathological immune response and thus might induce a beneficial effect [159]. The experience in the TPE treatment of MS exclusively involves the use of nanomembrane-based TPE (Figure 6) [17]. This innovative method involves passage of the patient’s blood through nanomembranes designed to filter out definite molecules [18][142][160][161][162][163][164][18,142,160,161,162,163,164]. There are pores in the nanomembrane with a diameter of 30–50 nm, which allow the filtration of molecules with a molecular weight below 40 kDa. The device has a volume of up to 70 mL [165]. The most widely used replacement is saline (NaCl 0.9), which is cheap and has no adverse effects, even when 25% of the circulating plasma is replaced [162]. The use of saline to replace the removed plasma is in agreement with the so-called low-volume plasma exchange (LVPE), which ranges from 350 mL to 2 l plasma volume removal for each separate procedure [166][167][168][169][166,167,168,169].