1. Basic Anatomy and Function of Retinal Ganglion Cells
RGCs play a crucial role in transmitting visual stimuli from the retina to the brain. From a morphological standpoint, RGCs consist of a cellular body, a complex dendritic structure, and a single axon. The primary function of dendrites is to receive synaptic inputs from bipolar and amacrine cells, which are intermediary neurons responsible for acquiring signals generated by photoreceptors
[1][2][3] (
Figure 1). The presence of hierarchical circuitry allows RGCs to engage in intricate signal integration and processing, leading to the conversion of photoreceptor responses into action potentials. The action potentials propagate through the RGC axons, which together constitute the optic nerve. They travel through the optic disc and optic canal to establish synaptic connections in the lateral geniculate nucleus (LGN), located in the thalamus
[2]. The visual information is transmitted from the LGN to the primary visual cortex (V1) for the purpose of undergoing higher-level processing and integration. Therefore, RGCs play a crucial role in connecting the initial light detection in the retina to the following brain visual perception.
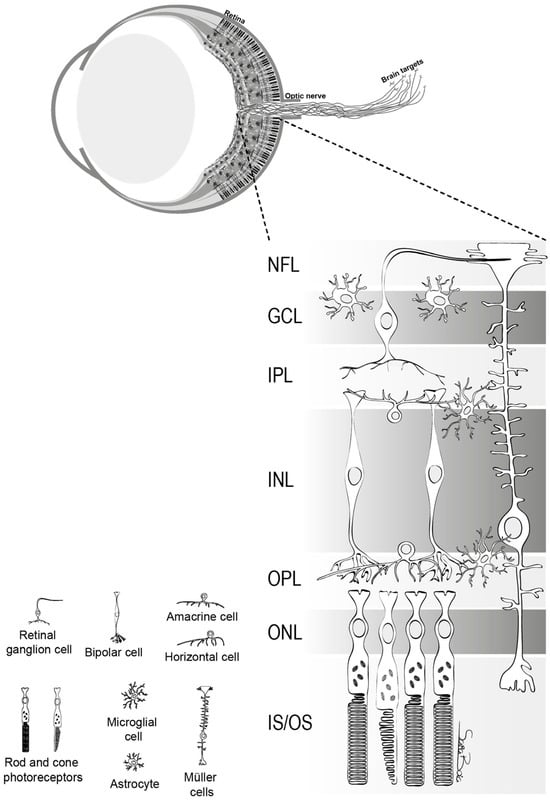
Figure 1. The structure of retina. This figure delineates the stratified architecture of the neural sensory retina, illustrating the cellular arrangement using distinct nuclear and plexiform layers. Photoreceptor nuclei, specifically those of rods and cones, reside within the outer nuclear layer (ONL), while the nuclei of various interneurons, including amacrine, bipolar, and horizontal cells, are primarily situated in the inner nuclear layer (INL). Retinal ganglion cells (RGCs) inhabit the ganglion cell layer (GCL), with their axonal projections extending through the nerve fiber layer (NFL). The retina comprises two macroglial variants: Müller cells, traversing the entire retinal thickness, and astrocytes, localized within the GCL. Microglia are chiefly found in the inner retinal regions and the outer plexiform layer (OPL)
[1].
In the human retina, a variety of RGCs are present, each specializing in distinct visual functions. The presence of heterogeneity among RGCs is a fundamental quality, as these cells can be differentiated into multiple subtypes based on their distinct morphological and physiological characteristics. Midget RGCs (P-Cell), comprising approximately 80% of RGCs, are primarily associated with the processing of visual information at a high level of detail and the discriminating of colors
[4]. Parasol RGCs (M-Cell), distinct from midget cells, are known to play a role in the detection of motion and changes in luminance
[5]. Midget RGCs receive inputs primarily from single cone photoreceptors and exhibit a sustained response to stimuli; the axons of midget RGCs project to dorsal parvocellular layers of the LGN. Parasol RGCs exhibit a larger dendritic field compared to midget RGCs and have a transient response to visual stimuli; the axons of parasol RGCs project to the ventral magnocellular layers of the RGC
[6]. Small bistratified RGCs, which constitute about 5–8% of the total, are an additional prominent subtype that receives synaptic inputs from both ON and OFF bipolar cells, allowing them to integrate signals from both pathways and contribute to the encoding of color contrast, particularly along the blue–yellow axis. These cells are components of the koniocellular pathway, implicated in color processing and integrating diverse visual features
[4][7]. Smooth monostratified RGCs, divided into ON and OFF cells, can relay spatial information in a complex manner
[8]. Melanopsin-containing intrinsically photosensitive RGCs (ipRGCs), essential for non-visual functions, represent about 1% of the total RGC population
[9]. Additionally, there are miscellaneous RGCs that defy existing classifications, underscoring the retina’s intricate complexity
[10]. Each specific subtype of RGCs makes distinct contributions to many areas of visual perception, encompassing spatial resolution, contrast sensitivity, and chromatic discrimination
[11][12]. Furthermore, it is important to note that certain subtypes of RGCs exhibit varying levels of vulnerability to degenerative alterations in glaucomatous circumstances. For example, in a pressure-induced environment, midget and parasol cells undergo degeneration that begins with dendritic arbor and concludes with cell soma shrinkage
[6]. As a result, it is crucial to develop a comprehensive understanding of the distinct functions and contributions of these subtypes.
2. Pathophysiology of Glaucoma and Current Theories on Retinal Ganglion Cell Vulnerability and Functionality in Glaucoma
Glaucoma is a heterogeneous group of ocular disorders that culminates in RGC degeneration and visual field loss. The mechanism underlying glaucomatous optic neuropathy is multifaceted, with theories on RGC vulnerability that include mechanical, vascular, excitatory, neurotrophic factor deprivation, and immune and inflammation mechanisms (
Table 1)
[13].
Table 1. Overview of glaucoma pathophysiology and contemporary theoretical frameworks.
| Theories |
Key Points |
| Mechanical theory |
- -
-
Mechanical strain, elevated IOP 1, and stiffness of sclera influence deformation of the lamina cribrosa (the most susceptible region in a pressurized eye).
- -
-
The sclera and lamina cribrosa act as load-bearing tissues of the optic nerve head.
|
| Vascular theory |
- -
-
Vascular insufficiency can cause RGC 2 dysfunction and death.
- -
-
Regulation for ocular blood flow is affected by OPP 3, temperature, and neural function.
- -
-
Vascular autoregulation and neurovascular coupling can be compromised in glaucoma.
- -
-
Mediators such as endothelin-1 and nitric oxide can affect blood flow and RGC health.
|
| Excitotoxicity theory |
- -
-
Excitotoxicity involves excessive glutamate release, leading to overstimulation of NMDA 4 receptors on RGCs, resulting in calcium influx, oxidative stress, mitochondrial dysfunction, and RGC injury.
|
| Neurotrophic factor deprivation theory |
- -
-
Interruption of axonal transport leads to reduced neurotrophic factors essential for RGC survival.
- -
-
BDNF 5, produced in the retina, is mediated through the TrkB 6 receptor, and its activation has a significant impact on RGC survival. Elevated IOP may hinder BDNF transport, resulting in RGC apoptosis and loss.
- -
-
Other neurotrophic factors linked to RGC health: CNTF 7, NGF 8, and GDNF 9.
|
| Oxidative Stress and Inflammation |
- -
-
Elevated IOP, ischemia-reperfusion injury, enzymatic degradation of neurotransmitters, and mitochondrial dysfunction contribute to ROS 10 overproduction and inflammatory mediators and can exacerbate RGC death.
- -
-
The trabecular meshwork is particularly vulnerable to oxidative damage.
- -
-
Elevated IOP can activate innate immune responses with increased microglial activity and complement expression.
- -
-
Adaptive immunity in glaucoma has been observed with induction of HSP-specific 11 memory T cells.
|
2.1. Mechanical Theories
The pathophysiology of glaucoma is complex, involving both mechanical and vascular components. Mechanical strain and compressive forces are exerted at the lamina cribrosa, recognized as the most susceptible region of a pressurized eye, subsequently affecting the structural stability and functionality of RGC axons
[14]. Research showed that elevated IOP from 5 to 50 mmHg over 24 h resulted in an average posterior deformation of the central lamina measuring 79 μm in human donor eyes and posterior displacement of the central lamina of 12 μm in response to an acute increase of IOP from 10 to 25 mmHg in human eyes
[15][16]. In addition, connective tissue of the peripapillary sclera and scleral canal wall are also responsible for bearing the forces generated by the IOP. The structural stiffness of the sclera plays a significant role in determining the deformation of the lamina cribrosa. Compliant sclera enables expansion of the scleral canal in response to elevated IOP, which results in tightening of the laminar beams within the canal and increases lamina resistance against posterior deformation. On the other hand, a rigid sclera exhibits limited capacity for canal expansion, hence necessitating the sole reliance on the structural rigidity of the lamina cribrosa to withstand the stress associated with elevated IOP
[16]. Therefore, the sclera and lamina cribrosa serve as the load-bearing tissues of the optic nerve head
[17].
2.2. Vascular Theories
The aforementioned discussion on mechanical theory suggests axonal compression at the lamina cribrosa, hindrance of axoplasmic flow, and disruption of retrograde neurotrophin transport to RGCs, ultimately resulting in cellular demise. Another aspect of RGC vulnerability is vascular insufficiency, where it is hypothesized that impaired perfusion pressure, disruption of vascular autoregulation, and loss of neurovascular coupling can lead to RGC dysfunction and death. In a study conducted by Shiga et al., the results of laser flow speckle flowgraphy indicated a statistically significant decrease in ocular blood flow in patients with preperimetric glaucoma compared to normal subjects
[18].
Ocular perfusion pressure (OPP) is defined as the sum of the systolic arterial blood pressure and one third of the difference between the systolic and diastolic pressures, minus the IOP. OPP signifies the oxygen supply and blood flow to the optic nerve; thus, it has been hypothesized that a reduction in OPP could increase the optic disc’s susceptibility to damage, thereby increasing the likelihood of glaucoma progression or development
[19]. Many studies have revealed a positive correlation between a reduction in OPP and an elevated prevalence of open-angle glaucoma
[20][21][22]. Additionally, previous research utilizing microspheres indicated a specific reduction of volume flow within the prelaminar and anterior laminar capillary beds when the OPP fell below 30 mm Hg
[23][24]. Regulation of ocular blood flow is affected by OPP, temperature, and neural function, and blood flow to the optic nerve head is mainly regulated by endothelial cells and circulating hormones
[25].
Vascular autoregulation refers to the regulatory mechanism that counteracts fluctuations in perfusion pressure, functioning optimally within a specific range of perfusion pressure. This mechanism can be compromised in conditions like glaucoma as well as other diseases, including diabetes mellitus. Various factors are involved in autoregulation, such as metabolic, myogenic, and neurogenic components. Direct autoregulation of ocular blood flow is difficult to assess, and many techniques have been used to measure ocular circulation, such as color Doppler imaging, scanning laser Doppler flowmeters, and optical coherence tomography (OCT). A study revealed individuals with glaucoma exhibited inadequate compensatory reaction of flow velocities in the short posterior ciliary artery (SPCA) to alterations in body position compared with healthy individuals, suggesting inadequate autoregulatory control may serve as a contributing factor to glaucoma pathogenesis
[26].
The central nervous system has a strong correlation between neuronal activity and blood flow, indicating a high level of coordination. Neurovascular coupling is the phenomenon wherein heightened neural activity leads to a concomitant augmentation in blood flow to the appropriate region
[27]. A study demonstrated a lower cerebral blood flow and functional connectivity strength coupling in glaucoma patients compared to controls, and the reduced ratio was significantly correlated with visual field defects and glaucoma stage, suggesting impaired neurovascular coupling in glaucoma patients
[28].
Numerous mediators orchestrate the regulation of ocular blood flow in glaucoma, which is a critical determinant of the health of the RGC and optic nerve head. Central among these mediators is endothelin-1 (ET-1), a potent vasoconstrictive peptide, which is elevated in aqueous and plasma concentrations of glaucoma patients
[29][30][31][32][33]. Clinical studies revealed worsening visual fields have higher plasma ET-1 compared to normal visual fields
[31], and animal studies demonstrated that low doses of ET-1 can induce glaucomatous changes in primates
[34]. Increased ET-1 levels result in an increase in IOP, which decreases ocular blood flow and astrocyte proliferation; consequently, this may ultimately lead to the degeneration of RGCs
[35]. A recent study utilized advanced imaging techniques, specifically OCT and OCT angiography, to assess vessel density in the peripapillary and macular regions of patients with glaucoma, revealing an inverse relationship with systemic ET-1 concentrations
[33]. Multivariate analyses indicated that IOP plays a less predictive role in the diminishment of retinal blood flow when compared to ET-1 levels in a glaucoma context. Elevated peripheral ET-1 has been identified as a potential risk indicator for monitoring vascular alterations in the optic nerve head of eyes affected by glaucoma. Conversely, nitric oxide acts as a pivotal vasodilator by stimulating cyclic guanine monophosphate (cGMP) to reduce intracellular calcium and thereby relaxation of smooth muscle cells and pericytes
[36]. In glaucoma patients, nitric oxide availability is reduced, which can cause a disruption in the equilibrium between vasoconstriction and vasodilation and ultimately results in a reduction of blood flow to the optic nerve head
[37].
2.3. Excitotoxicity Theory
Research into the role of excitotoxicity, which is mediated by excessive glutamate release and subsequent overstimulation of N-methyl-d-aspartate (NMDA) receptors on RGCs, has revealed the molecular and cellular pathways that contribute to RGC injury in glaucoma. Glutamate serves as a primary neurotransmitter inside the retinal network, facilitating communication between the photoreceptors, bipolar cells, and RGCs
[38]. The process of glutamate neurotransmission is intricately regulated by inhibitory and modulatory neurotransmitters, including γ-aminobutyric acid (GABA), glycine, acetylcholine, dopamine, serine, substance P, and several neuropeptides
[39]. Excessive glutamate release causes overactivation of the NMDA receptor, which induces calcium influx, oxidative stress, and mitochondrial dysfunction, all of which contribute to the degeneration of RGCs. An increase in glutamate has not been consistently seen in retinal pathologies such as glaucoma
[40]; however, due to the progressive nature of glaucoma, glutamate levels may not significantly increase acutely or accumulate during the course of the disease and may only rise in specific parts of the retina or optic nerve.
Generally, when glutamate is released, the initial depolarization of the cell membrane through α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) receptor activation leads to an elevation in the likelihood of NMDA receptor opening. This triggers a transient influx of calcium ions into the intracellular space. Calcium can also be released via the inositol 1,4,5-trisphosphate receptors (IP3R) and the ryanodine receptors (RyR) that are present on the membrane of the endoplasmic reticulum. Functional mitochondrial metabolism plays a crucial role in maintaining intracellular calcium homeostasis through the reuptake of calcium by the Sarco-Endoplasmic Reticulum Calcium ATPase (SERCA) and the extrusion of calcium to the extracellular space via the sodium-calcium ATPase. Furthermore, the extrusion of calcium is facilitated by the sodium-calcium exchanger (NCX) through the utilization of the physiological sodium gradient. The failure of RGCs to adequately regulate or remove excessive calcium ions disrupts the homeostatic mechanisms, which ultimately culminates in cellular demise. The involvement of NCX in the process of retinal cell death triggered by NMDA and ischemia-reperfusion has been suggested
[41][42]. High levels of intracellular calcium along with heightened activity of catabolic enzymes can trigger a cascade of events, leading to RGC apoptosis and necrosis
[43][44]. The subsequent cascade encompasses mitochondrial membrane depolarization, activation of caspases, generation of harmful oxygen and nitrogen free radicals, and manifestation of cellular toxicity.
Normal glutamate excitotoxicity is minimized by its quick absorption by glutamate transporters in glial cells surrounding RGCs. The majority of glutamate transporters in the retina are located at the synaptic cleft in order to quickly remove released glutamate and prevent overflow
[45]. Glutamate uptake by Müller cells via glutamate/aspartate transporters (GLAST or EAAT1) is crucial for maintaining physiological glutamate levels
[46]. Retinal glia and neurons produce several glutamate transporters, such as GLT-1 (EAAT2), excitatory amino acid carrier 1 (EAAC1 in rats or EAAT3 in humans), EAAT4, and EAAT5 in rats. Decreased retinal GLAST has been correlated with glaucoma in rats and mice
[47][48], and GLAST-deficient mice displayed loss of RGCs
[49].
The profound vulnerability of RGCs is underscored by the imbalance between neurotransmitter release and uptake, as well as the cascade of intracellular events triggered by excessive receptor stimulation.
2.4. Neurotrophic Factor Deprivation Theory
According to the neurotrophic factor deprivation theory, the interruption of axonal transport results in a diminished supply of neurotrophic factors, which are essential for the survival of RGCs. Neurotrophins, which are diffusible trophic molecules, enhance the survival of mature neurons in the central nervous system (CNS), which degenerate in response to an extensive range of stimuli. Brain-derived neurotrophic factor (BDNF) has garnered significant interest within the field of neurotrophins due to its influential impact on the survival of RGCs. The initial evidence of BDNF’s neuroprotective properties in RGCs occurred in 1986
[50]. BDNF is produced locally in the retina by RGCs and glia, but it is also strongly expressed in the superior colliculus, where it is retrogradely transported to the optic nerve head and to the cell bodies of RGCs
[45][51]. The effects of BDNF are mediated through tropomyosin receptor kinase B (TrkB) on the presynaptic cell surface, and TrkB activation leads to three major signaling cascades (mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and phospholipase Cγ (PLC-γ)), which are responsible for promoting the survival of RGCs
[52]. Elevated IOP and the resulting mechanical strain on the optic nerve head may hinder the retrograde transport of BDNF in glaucoma. When the stressed RGC is deprived of BDNF, Jun-N terminal kinase (JNK)-mediated signaling may be triggered, which may activate the proapoptotic BCL-2 family of proteins and cause mitochondrial dysfunction
[51][53][54]. Numerous studies have demonstrated a correlation between RGC loss and impaired BDNF-TrkB signaling
[55][56][57]. Consistent clinically, BDNF deficit was demonstrated in serum, aqueous humor, and lacrimal tears of patients with early glaucomatous change
[58][59]. Apart from BDNF, additional neurotrophic factors have been linked to RGC health, including ciliary neurotrophic factor (CNTF), nerve growth factor (NGF), and glial cell line–derived neurotrophic factor (GDNF)
[53].
The multifaceted nature of neurotrophic factors in the retina emphasizes the importance of its support in RGC health.
2.5. Oxidative Stress and Inflammation
Oxidative stress and neuroinflammation are implicated in the pathogenesis of glaucoma, where reactive oxygen species (ROS) and inflammatory mediators precipitate cellular damage and contribute to an excitotoxic environment, thereby exacerbating RGC death. Multiple factors can contribute to increased ROS production in glaucomatous eyes. Ischemia-reperfusion injury, elevated IOP, and mitochondrial dysfunction are among the principal factors that contribute to the overproduction of ROS in the optic nerve head and retina
[45][60]. The principal roles of mitochondria encompass the synthesis of adenosine triphosphate (ATP) via the oxidative phosphorylation pathway and the orchestration of apoptotic cell death
[61]. ROS are consistently generated within mitochondria via the electron transport chain. However, enzymatic degradation of neurotransmitters, neuroinflammatory mediators, and redox reactions can also contribute to ROS production
[62]. In the anterior chamber, the trabecular meshwork (TM) is the most susceptible tissue to oxidative injury
[63]. Oxidative stress on the TM can induce numerous detrimental effects, including altering cell-cycle progression
[64][65], changes in the extracellular matrix
[66], rearrangement of TM cell cytoskeletal structures, loss of cell-matrix adhesion
[67], and alteration of membrane permeability
[68]. Additionally, it can induce inflammatory cytokine release and initiate RGC apoptosis
[45]. Mitochondrial dysfunction augments the production of ROS while concurrently diminishing the synthesis of ATP. Neuronal cells, which demand substantial energy to sustain electrochemical gradients crucial for signal conduction, are exceedingly susceptible to perturbations in mitochondrial function. The generation of ATP predominantly occurs within the mitochondria via the oxidative phosphorylation mechanism of the electron transport chain, complemented by the glycolytic process
[69]. Defects in mitochondrial complex 1 were found to induce ROS release and decrease ATP levels in human glaucomatous TM cells
[70]; a study found that RGC death occurred 3–4 months after elevated IOP, with an 18% reduction in mitochondrial membrane potential
[71]. Dynamin-related protein 1 (Drp1), which is present on the outer mitochondrial membrane, has been shown to play a role in glaucoma
[72]. A recent study showed that the ERK1/2-Drp1-ROS axis can cause mitochondrial dysfunction and apoptosis in RGCs
[73]. The involvement of mitochondria in glial neuroinflammation processes is facilitated by the activation of NF-κB through the generation of mitochondrial ROS, resulting in the production of inflammatory cytokines
[74].
In response to cellular injury and oxidative stress, numerous pro-inflammatory mediators are upregulated. Normally, the eye is regarded as an immune-privileged site; however, the permeability of the blood–retina barrier may be altered by factors such as oxidative stress, vascular endothelial growth factor, and inflammation
[75]. In addition to chemokines and adhesion molecules, cytokines such as interleukins 1β (IL-1β) and tumor necrosis factor-alpha (TNF-α) exhibit increased levels in glaucomatous tissues
[76]. Elevated IOP can trigger an innate immune response with researchers finding an increase in microglia activity and cell density as well as expression of the complement of C1q in the retina and optic nerve in mice with glaucoma
[77][78]. Adaptive immunity of glaucoma was seen when heat shock protein (HSP)-specific memory T cells were induced by commensal microflora
[79]. HSP-27, HSP-60, and HSP-70 autoantibodies have been found in the sera of glaucoma patients
[80]. In addition, microglial cells are activated in response to elevated IOP by producing cytokines, mediators, and enzymes that can alter the ECM
[81]. Microglial cells can exhibit two phenotypes, M1 (pro-inflammatory) or M2 (neuroprotective); however, in glaucomatous eyes, the majority of microglia do not show the M2 phenotype, and the activities of microglia may have detrimental consequences for the glaucomatous optic disc
[82].
In conclusion, the RGC vulnerability hypotheses in glaucoma are multifaceted, encompassing mechanical, vascular, metabolic, excitotoxic, and inflammatory dimensions. The combination of these theories highlights the complexity of glaucomatous neurodegeneration and necessitates a holistic understanding of the changes in RGC functionality, thereby guiding the development of nuanced therapeutic strategies aimed at reducing RGC vulnerability and preserving visual function.
 +1 credit
+1 credit