Glaucoma, a leading cause of irreversible blindness globally, primarily affects retinal ganglion cells (RGCs). Glaucoma is a multifactorial eye disease defined by the progressive degeneration of retinal ganglion cells (RGCs) and their axons, eventually leading to irreversible vision loss. It has been projected that the worldwide prevalence of individuals affected by glaucoma will experience a substantial growth of 74% between the years 2013 and 2040. Glaucoma poses significant clinical and public health challenges as one of the leading causes of blindness around the globe. While elevated intraocular pressure (IOP) remains a significant risk factor and therapeutic target for glaucoma, it is becoming increasingly apparent that other factors may play a role in the disease’s pathogenesis and progression. The structure and function of RGCs, which serve as the ultimate output neurons of the retina and transmit visual information to the brain, are among the most crucial components being studied.
- retinal ganglion cells
- glaucoma
- neuroprotection
- neurodegeneration
- optical coherence tomography
- imaging
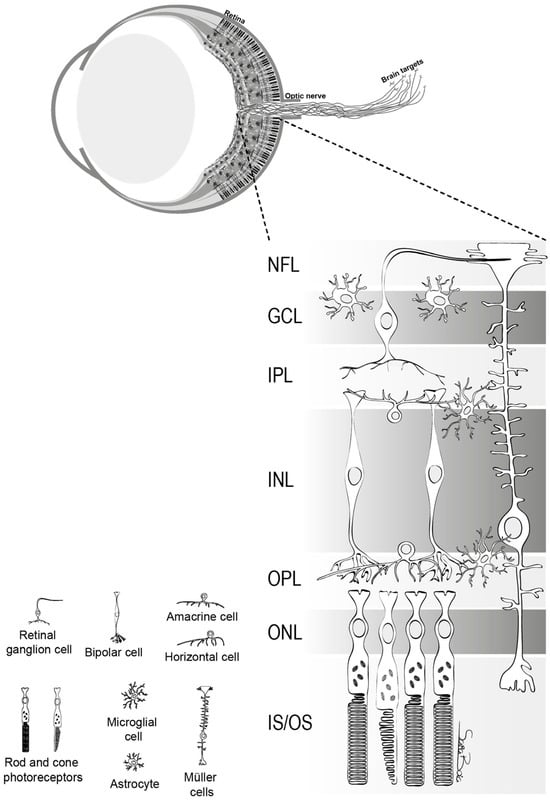
1. Basic Anatomy and Function of Retinal Ganglion Cells
2. Pathophysiology of Glaucoma and Current Theories on Retinal Ganglion Cell Vulnerability and Functionality in Glaucoma
| Theories | Key Points |
|---|---|
| Mechanical theory |
|
| Vascular theory |
|
| Excitotoxicity theory |
|
| Neurotrophic factor deprivation theory |
|
| Oxidative Stress and Inflammation |
|
2.1. Mechanical Theories
2.2. Vascular Theories
2.3. Excitotoxicity Theory
2.4. Neurotrophic Factor Deprivation Theory
2.5. Oxidative Stress and Inflammation
This entry is adapted from the peer-reviewed paper 10.3390/cells12242797
References
- Boia, R.; Ruzafa, N.; Aires, I.D.; Pereiro, X.; Ambrosio, A.F.; Vecino, E.; Santiago, A.R. Neuroprotective Strategies for Retinal Ganglion Cell Degeneration: Current Status and Challenges Ahead. Int. J. Mol. Sci. 2020, 21, 2262.
- Vernazza, S.; Oddone, F.; Tirendi, S.; Bassi, A.M. Risk Factors for Retinal Ganglion Cell Distress in Glaucoma and Neuroprotective Potential Intervention. Int. J. Mol. Sci. 2021, 22, 7994.
- Masland, R.H. The neuronal organization of the retina. Neuron 2012, 76, 266–280.
- Martin, P.R. Colour processing in the primate retina: Recent progress. J. Physiol. 1998, 513 Pt 3, 631–638.
- Manookin, M.B.; Patterson, S.S.; Linehan, C.M. Neural Mechanisms Mediating Motion Sensitivity in Parasol Ganglion Cells of the Primate Retina. Neuron 2018, 97, 1327–1340.e1324.
- Weber, A.J.; Kaufman, P.L.; Hubbard, W.C. Morphology of single ganglion cells in the glaucomatous primate retina. Invest. Ophthalmol. Vis. Sci. 1998, 39, 2304–2320.
- Martin, P.R.; Grunert, U. Analysis of the short wavelength-sensitive (“blue”) cone mosaic in the primate retina: Comparison of New World and Old World monkeys. J. Comp. Neurol. 1999, 406, 1–14.
- Crook, J.D.; Peterson, B.B.; Packer, O.S.; Robinson, F.R.; Gamlin, P.D.; Troy, J.B.; Dacey, D.M. The smooth monostratified ganglion cell: Evidence for spatial diversity in the Y-cell pathway to the lateral geniculate nucleus and superior colliculus in the macaque monkey. J. Neurosci. 2008, 28, 12654–12671.
- Do, M.T.H. Melanopsin and the Intrinsically Photosensitive Retinal Ganglion Cells: Biophysics to Behavior. Neuron 2019, 104, 205–226.
- Sanes, J.R.; Masland, R.H. The types of retinal ganglion cells: Current status and implications for neuronal classification. Annu. Rev. Neurosci. 2015, 38, 221–246.
- Silveira, L.C.; Saito, C.A.; Lee, B.B.; Kremers, J.; da Silva Filho, M.; Kilavik, B.E.; Yamada, E.S.; Perry, V.H. Morphology and physiology of primate M- and P-cells. Prog. Brain Res. 2004, 144, 21–46.
- Paknahad, J.; Loizos, K.; Yue, L.; Humayun, M.S.; Lazzi, G. Color and cellular selectivity of retinal ganglion cell subtypes through frequency modulation of electrical stimulation. Sci. Rep. 2021, 11, 5177.
- Flammer, J.; Mozaffarieh, M. What is the present pathogenetic concept of glaucomatous optic neuropathy? Surv. Ophthalmol. 2007, 52 (Suppl. 2), S162–S173.
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: A review. JAMA 2014, 311, 1901–1911.
- Yan, D.B.; Coloma, F.M.; Metheetrairut, A.; Trope, G.E.; Heathcote, J.G.; Ethier, C.R. Deformation of the lamina cribrosa by elevated intraocular pressure. Br. J. Ophthalmol. 1994, 78, 643–648.
- Downs, J.C.; Roberts, M.D.; Burgoyne, C.F. Mechanical environment of the optic nerve head in glaucoma. Optom. Vis. Sci. 2008, 85, 425–435.
- Burgoyne, C.F.; Downs, J.C.; Bellezza, A.J.; Suh, J.K.; Hart, R.T. The optic nerve head as a biomechanical structure: A new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog. Retin. Eye Res. 2005, 24, 39–73.
- Shiga, Y.; Kunikata, H.; Aizawa, N.; Kiyota, N.; Maiya, Y.; Yokoyama, Y.; Omodaka, K.; Takahashi, H.; Yasui, T.; Kato, K.; et al. Optic Nerve Head Blood Flow, as Measured by Laser Speckle Flowgraphy, Is Significantly Reduced in Preperimetric Glaucoma. Curr. Eye Res. 2016, 41, 1447–1453.
- Caprioli, J.; Coleman, A.L.; Blood Flow in Glaucoma, D. Blood pressure, perfusion pressure, and glaucoma. Am. J. Ophthalmol. 2010, 149, 704–712.
- Leske, M.C.; Wu, S.Y.; Hennis, A.; Honkanen, R.; Nemesure, B.; Group, B.E.S. Risk factors for incident open-angle glaucoma: The Barbados Eye Studies. Ophthalmology 2008, 115, 85–93.
- Memarzadeh, F.; Ying-Lai, M.; Chung, J.; Azen, S.P.; Varma, R.; Los Angeles Latino Eye Study, G. Blood pressure, perfusion pressure, and open-angle glaucoma: The Los Angeles Latino Eye Study. Invest. Ophthalmol. Vis. Sci. 2010, 51, 2872–2877.
- Zheng, Y.; Wong, T.Y.; Mitchell, P.; Friedman, D.S.; He, M.; Aung, T. Distribution of ocular perfusion pressure and its relationship with open-angle glaucoma: The singapore malay eye study. Invest. Ophthalmol. Vis. Sci. 2010, 51, 3399–3404.
- Geijer, C.; Bill, A. Effects of raised intraocular pressure on retinal, prelaminar, laminar, and retrolaminar optic nerve blood flow in monkeys. Invest. Ophthalmol. Vis. Sci. 1979, 18, 1030–1042.
- Alm, A.; Bill, A. Ocular and optic nerve blood flow at normal and increased intraocular pressures in monkeys (Macaca irus): A study with radioactively labelled microspheres including flow determinations in brain and some other tissues. Exp. Eye Res. 1973, 15, 15–29.
- Grieshaber, M.C.; Mozaffarieh, M.; Flammer, J. What is the link between vascular dysregulation and glaucoma? Surv. Ophthalmol. 2007, 52 (Suppl. 2), S144–S154.
- Galambos, P.; Vafiadis, J.; Vilchez, S.E.; Wagenfeld, L.; Matthiessen, E.T.; Richard, G.; Klemm, M.; Zeitz, O. Compromised autoregulatory control of ocular hemodynamics in glaucoma patients after postural change. Ophthalmology 2006, 113, 1832–1836.
- Wang, X.; Wang, M.; Liu, H.; Mercieca, K.; Prinz, J.; Feng, Y.; Prokosch, V. The Association between Vascular Abnormalities and Glaucoma-What Comes First? Int. J. Mol. Sci. 2023, 24, 13211.
- Wang, Q.; Qu, X.; Chen, W.; Wang, H.; Huang, C.; Li, T.; Wang, N.; Xian, J. Altered coupling of cerebral blood flow and functional connectivity strength in visual and higher order cognitive cortices in primary open angle glaucoma. J. Cereb. Blood Flow. Metab. 2021, 41, 901–913.
- Ghanem, A.A.; Elewa, A.M.; Arafa, L.F. Endothelin-1 and nitric oxide levels in patients with glaucoma. Ophthalmic Res. 2011, 46, 98–102.
- Shoshani, Y.Z.; Harris, A.; Shoja, M.M.; Rusia, D.; Siesky, B.; Arieli, Y.; Wirostko, B. Endothelin and its suspected role in the pathogenesis and possible treatment of glaucoma. Curr. Eye Res. 2012, 37, 1–11.
- Emre, M.; Orgul, S.; Haufschild, T.; Shaw, S.G.; Flammer, J. Increased plasma endothelin-1 levels in patients with progressive open angle glaucoma. Br. J. Ophthalmol. 2005, 89, 60–63.
- McGrady, N.R.; Minton, A.Z.; Stankowska, D.L.; He, S.; Jefferies, H.B.; Krishnamoorthy, R.R. Upregulation of the endothelin A (ETA) receptor and its association with neurodegeneration in a rodent model of glaucoma. BMC Neurosci. 2017, 18, 27.
- Lommatzsch, C.; Rothaus, K.; Schopmeyer, L.; Feldmann, M.; Bauer, D.; Grisanti, S.; Heinz, C.; Kasper, M. Elevated endothelin-1 levels as risk factor for an impaired ocular blood flow measured by OCT-A in glaucoma. Sci. Rep. 2022, 12, 11801.
- Orgul, S.; Cioffi, G.A.; Bacon, D.R.; Van Buskirk, E.M. An endothelin-1-induced model of chronic optic nerve ischemia in rhesus monkeys. J. Glaucoma 1996, 5, 135–138.
- Wang, L.; Fortune, B.; Cull, G.; Dong, J.; Cioffi, G.A. Endothelin B receptor in human glaucoma and experimentally induced optic nerve damage. Arch. Ophthalmol. 2006, 124, 717–724.
- Haefliger, I.O.; Dettmann, E.; Liu, R.; Meyer, P.; Prunte, C.; Messerli, J.; Flammer, J. Potential role of nitric oxide and endothelin in the pathogenesis of glaucoma. Surv. Ophthalmol. 1999, 43 (Suppl. 1), S51–S58.
- Toda, N.; Nakanishi-Toda, M. Nitric oxide: Ocular blood flow, glaucoma, and diabetic retinopathy. Prog. Retin. Eye Res. 2007, 26, 205–238.
- Masland, R.H. The fundamental plan of the retina. Nat. Neurosci. 2001, 4, 877–886.
- Boccuni, I.; Fairless, R. Retinal Glutamate Neurotransmission: From Physiology to Pathophysiological Mechanisms of Retinal Ganglion Cell Degeneration. Life 2022, 12, 638.
- Lotery, A.J. Glutamate excitotoxicity in glaucoma: Truth or fiction? Eye 2005, 19, 369–370.
- Inokuchi, Y.; Shimazawa, M.; Nakajima, Y.; Komuro, I.; Matsuda, T.; Baba, A.; Araie, M.; Kita, S.; Iwamoto, T.; Hara, H. A Na+/Ca2+ exchanger isoform, NCX1, is involved in retinal cell death after N-methyl-D-aspartate injection and ischemia-reperfusion. J. Neurosci. Res. 2009, 87, 906–917.
- Brittain, M.K.; Brustovetsky, T.; Sheets, P.L.; Brittain, J.M.; Khanna, R.; Cummins, T.R.; Brustovetsky, N. Delayed calcium dysregulation in neurons requires both the NMDA receptor and the reverse Na+/Ca2+ exchanger. Neurobiol. Dis. 2012, 46, 109–117.
- Brandt, S.K.; Weatherly, M.E.; Ware, L.; Linn, D.M.; Linn, C.L. Calcium preconditioning triggers neuroprotection in retinal ganglion cells. Neuroscience 2011, 172, 387–397.
- Hartwick, A.T.; Hamilton, C.M.; Baldridge, W.H. Glutamatergic calcium dynamics and deregulation of rat retinal ganglion cells. J. Physiol. 2008, 586, 3425–3446.
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181.
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Muller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424.
- Martin, K.R.; Levkovitch-Verbin, H.; Valenta, D.; Baumrind, L.; Pease, M.E.; Quigley, H.A. Retinal glutamate transporter changes in experimental glaucoma and after optic nerve transection in the rat. Invest. Ophthalmol. Vis. Sci. 2002, 43, 2236–2243.
- Schuettauf, F.; Thaler, S.; Bolz, S.; Fries, J.; Kalbacher, H.; Mankowska, A.; Zurakowski, D.; Zrenner, E.; Rejdak, R. Alterations of amino acids and glutamate transport in the DBA/2J mouse retina; possible clues to degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 1157–1168.
- Harada, T.; Harada, C.; Nakamura, K.; Quah, H.M.; Okumura, A.; Namekata, K.; Saeki, T.; Aihara, M.; Yoshida, H.; Mitani, A.; et al. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J. Clin. Investig. 2007, 117, 1763–1770.
- Johnson, J.E.; Barde, Y.A.; Schwab, M.; Thoenen, H. Brain-derived neurotrophic factor supports the survival of cultured rat retinal ganglion cells. J. Neurosci. 1986, 6, 3031–3038.
- Dekeyster, E.; Geeraerts, E.; Buyens, T.; Van den Haute, C.; Baekelandt, V.; De Groef, L.; Salinas-Navarro, M.; Moons, L. Tackling Glaucoma from within the Brain: An Unfortunate Interplay of BDNF and TrkB. PLoS ONE 2015, 10, e0142067.
- Gonzalez-Hoyuela, M.; Barbas, J.A.; Rodriguez-Tebar, A. The autoregulation of retinal ganglion cell number. Development 2001, 128, 117–124.
- Kimura, A.; Namekata, K.; Guo, X.; Harada, C.; Harada, T. Neuroprotection, Growth Factors and BDNF-TrkB Signalling in Retinal Degeneration. Int. J. Mol. Sci. 2016, 17, 1584.
- Lambuk, L.; Mohd Lazaldin, M.A.; Ahmad, S.; Iezhitsa, I.; Agarwal, R.; Uskokovic, V.; Mohamud, R. Brain-Derived Neurotrophic Factor-Mediated Neuroprotection in Glaucoma: A Review of Current State of the Art. Front. Pharmacol. 2022, 13, 875662.
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest. Ophthalmol. Vis. Sci. 2000, 41, 764–774.
- Quigley, H.A.; McKinnon, S.J.; Zack, D.J.; Pease, M.E.; Kerrigan-Baumrind, L.A.; Kerrigan, D.F.; Mitchell, R.S. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest. Ophthalmol. Vis. Sci. 2000, 41, 3460–3466.
- Osborne, A.; Khatib, T.Z.; Songra, L.; Barber, A.C.; Hall, K.; Kong, G.Y.X.; Widdowson, P.S.; Martin, K.R. Neuroprotection of retinal ganglion cells by a novel gene therapy construct that achieves sustained enhancement of brain-derived neurotrophic factor/tropomyosin-related kinase receptor-B signaling. Cell Death Dis. 2018, 9, 1007.
- Igarashi, T.; Nakamoto, K.; Kobayashi, M.; Suzuki, H.; Arima, T.; Tobita, Y.; Takao, K.; Igarashi, T.; Okuda, T.; Okada, T.; et al. Brain-derived Neurotrophic Factor in the Aqueous Humor of Glaucoma Patients. J. Nippon. Med. Sch. 2021, 88, 128–132.
- Shpak, A.A.; Guekht, A.B.; Druzhkova, T.A.; Kozlova, K.I.; Gulyaeva, N.V. Brain-Derived Neurotrophic Factor in Patients with Primary Open-Angle Glaucoma and Age-related Cataract. Curr. Eye Res. 2018, 43, 224–231.
- Tezel, G. Multifactorial Pathogenic Processes of Retinal Ganglion Cell Degeneration in Glaucoma towards Multi-Target Strategies for Broader Treatment Effects. Cells 2021, 10, 1372.
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best. Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723.
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658.
- Izzotti, A.; Sacca, S.C.; Longobardi, M.; Cartiglia, C. Sensitivity of ocular anterior chamber tissues to oxidative damage and its relevance to the pathogenesis of glaucoma. Invest. Ophthalmol. Vis. Sci. 2009, 50, 5251–5258.
- Clopton, D.A.; Saltman, P. Low-level oxidative stress causes cell-cycle specific arrest in cultured cells. Biochem. Biophys. Res. Commun. 1995, 210, 189–196.
- Giancotti, F.G. Integrin signaling: Specificity and control of cell survival and cell cycle progression. Curr. Opin. Cell Biol. 1997, 9, 691–700.
- Knepper, P.A.; Goossens, W.; Hvizd, M.; Palmberg, P.F. Glycosaminoglycans of the human trabecular meshwork in primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci. 1996, 37, 1360–1367.
- Zhou, L.; Li, Y.; Yue, B.Y. Oxidative stress affects cytoskeletal structure and cell-matrix interactions in cells from an ocular tissue: The trabecular meshwork. J. Cell Physiol. 1999, 180, 182–189.
- Alvarado, J.A.; Alvarado, R.G.; Yeh, R.F.; Franse-Carman, L.; Marcellino, G.R.; Brownstein, M.J. A new insight into the cellular regulation of aqueous outflow: How trabecular meshwork endothelial cells drive a mechanism that regulates the permeability of Schlemm’s canal endothelial cells. Br. J. Ophthalmol. 2005, 89, 1500–1505.
- Kong, G.Y.X.; Van Bergen, N.J.; Trounce, I.A.; Crowston, J.G. Mitochondrial Dysfunction and Glaucoma. J. Glaucoma 2009, 18, 93–100.
- He, Y.; Leung, K.W.; Zhang, Y.H.; Duan, S.; Zhong, X.F.; Jiang, R.Z.; Peng, Z.; Tombran-Tink, J.; Ge, J. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: Protection by antioxidants. Invest. Ophthalmol. Vis. Sci. 2008, 49, 1447–1458.
- Mittag, T.W.; Danias, J.; Pohorenec, G.; Yuan, H.M.; Burakgazi, E.; Chalmers-Redman, R.; Podos, S.M.; Tatton, W.G. Retinal damage after 3 to 4 months of elevated intraocular pressure in a rat glaucoma model. Invest. Ophthalmol. Vis. Sci. 2000, 41, 3451–3459.
- Shi, W.; Tan, C.; Liu, C.; Chen, D. Mitochondrial fission mediated by Drp1-Fis1 pathway and neurodegenerative diseases. Rev. Neurosci. 2023, 34, 275–294.
- Zeng, Z.; You, M.; Fan, C.; Rong, R.; Li, H.; Xia, X. Pathologically high intraocular pressure induces mitochondrial dysfunction through Drp1 and leads to retinal ganglion cell PANoptosis in glaucoma. Redox Biol. 2023, 62, 102687.
- Tezel, G. Molecular regulation of neuroinflammation in glaucoma: Current knowledge and the ongoing search for new treatment targets. Prog. Retin. Eye Res. 2022, 87, 100998.
- Kaur, C.; Ling, E.A. Blood brain barrier in hypoxic-ischemic conditions. Curr. Neurovasc Res. 2008, 5, 71–81.
- Chua, J.; Vania, M.; Cheung, C.M.; Ang, M.; Chee, S.P.; Yang, H.; Li, J.; Wong, T.T. Expression profile of inflammatory cytokines in aqueous from glaucomatous eyes. Mol. Vis. 2012, 18, 431–438.
- Wei, X.; Cho, K.S.; Thee, E.F.; Jager, M.J.; Chen, D.F. Neuroinflammation and microglia in glaucoma: Time for a paradigm shift. J. Neurosci. Res. 2019, 97, 70–76.
- Howell, G.R.; Macalinao, D.G.; Sousa, G.L.; Walden, M.; Soto, I.; Kneeland, S.C.; Barbay, J.M.; King, B.L.; Marchant, J.K.; Hibbs, M.; et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J. Clin. Investig. 2011, 121, 1429–1444.
- Chen, H.; Cho, K.S.; Vu, T.H.K.; Shen, C.H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat. Commun. 2018, 9, 3209.
- Von Thun Und Hohenstein-Blaul, N.; Bell, K.; Pfeiffer, N.; Grus, F.H. Autoimmune aspects in glaucoma. Eur. J. Pharmacol. 2016, 787, 105–118.
- Yuan, L.; Neufeld, A.H. Activated microglia in the human glaucomatous optic nerve head. J. Neurosci. Res. 2001, 64, 523–532.
- de Hoz, R.; Gallego, B.I.; Ramirez, A.I.; Rojas, B.; Salazar, J.J.; Valiente-Soriano, F.J.; Aviles-Trigueros, M.; Villegas-Perez, M.P.; Vidal-Sanz, M.; Trivino, A.; et al. Rod-like microglia are restricted to eyes with laser-induced ocular hypertension but absent from the microglial changes in the contralateral untreated eye. PLoS ONE 2013, 8, e83733.