 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | AMER AL AL KHALIFA | -- | 4184 | 2023-12-14 16:12:01 | | | |
| 2 | Lindsay Dong | Meta information modification | 4184 | 2023-12-18 08:36:45 | | |
Video Upload Options
The blood–brain barrier (BBB) is a unique and selective feature of the central nervous system’s vasculature. BBB dysfunction has been observed as an early sign of Alzheimer’s Disease (AD) before the onset of dementia or neurodegeneration. The intricate relationship between the BBB and the pathogenesis of AD, especially in the context of neurovascular coupling and the overlap of pathophysiology in neurodegenerative and cerebrovascular diseases, underscores the urgency to understand the BBB’s role more deeply. Preserving or restoring the BBB function emerges as a potentially promising strategy for mitigating the progression and severity of AD. Molecular and genetic changes, such as the isoform ε4 of apolipoprotein E (ApoEε4), a significant genetic risk factor and a promoter of the BBB dysfunction, have been shown to mediate the BBB disruption. Additionally, receptors and transporters like the low-density lipoprotein receptor-related protein 1 (LRP1), P-glycoprotein (P-gp), and the receptor for advanced glycation end products (RAGEs) have been implicated in AD’s pathogenesis.
1. Introduction
2. NVU and the BBB
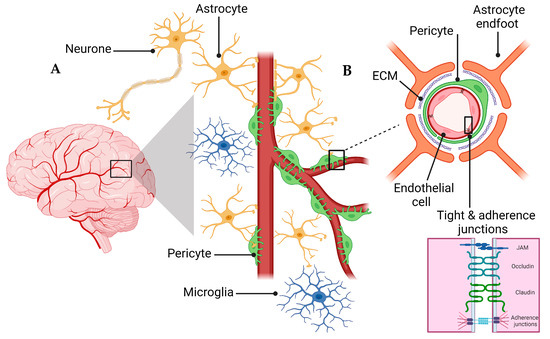
3. BBB Dysregulation with Aging
4. BBB Dysfunction in AD
4.1. Effect of Comorbidities on BBB
4.2. ApoE and BBB Dysfunction in AD
4.3. Tight Junctions’ Role in BBB Health and Disease
4.4. Transporters’ Role in BBB Health and Disease
5. BBB Breakdown Mechanisms
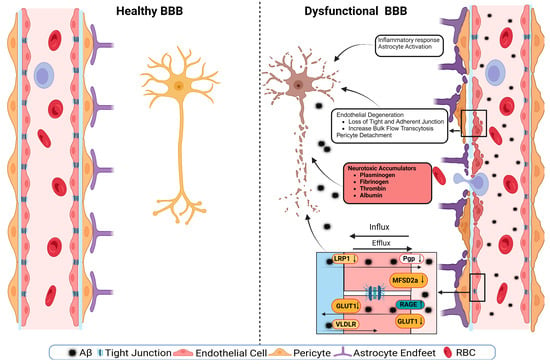
6. The BBB as a Therapeutic Target
References
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32.
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387.
- Alzheimer’s association. 2023 Alzheimer’s disease facts and figures. Alzheimers Dement 2023, 19, 1598–1695.
- FDA. FDA Grants Accelerated Approval for Alzheimer’s Disease Treatment. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment (accessed on 15 May 2023).
- FDA. FDA Grants Accelerated Approval for Alzheimer’s Drug. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug (accessed on 15 May 2023).
- Marucci, G.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352.
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189.
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.-d.-C.; Diaz-Cintra, S. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754.
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900.
- Li, S.; Wang, C.; Wang, Z.; Tan, J. Involvement of cerebrovascular abnormalities in the pathogenesis and progression of Alzheimer’s disease: An adrenergic approach. Aging Albany NY 2021, 13, 21791–21806.
- Ringman, J.M.; Sachs, M.C.; Zhou, Y.; Monsell, S.E.; Saver, J.L.; Vinters, H.V. Clinical predictors of severe cerebral amyloid angiopathy and influence of APOE genotype in persons with pathologically verified Alzheimer disease. JAMA Neurol. 2014, 71, 878–883.
- Theodorou, A.; Tsantzali, I.; Kapaki, E.; Constantinides, V.C.; Voumvourakis, K.; Tsivgoulis, G.; Paraskevas, G.P. Cerebrospinal fluid biomarkers and apolipoprotein E genotype in cerebral amyloid angiopathy. A narrative review. Cereb. Circ.-Cogn. Behav. 2021, 2, 100010.
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118.
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303.
- Salloway, S.; Correia, S.; Peck, J.; Harrington, C. Dementia with Lewy bodies: A diagnostic and treatment challenge. Med. Health Rhode Isl. 2002, 85, 207–209.
- Montagne, A.; Nikolakopoulou, A.M.; Huuskonen, M.T.; Sagare, A.P.; Lawson, E.J.; Lazic, D.; Rege, S.V.; Grond, A.; Zuniga, E.; Barnes, S.R. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-β. Nat. Aging 2021, 1, 506–520.
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201.
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53.
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and dysfunction of the blood-brain barrier. Cell 2015, 163, 1064–1078.
- Guttenplan, K.A.; Liddelow, S.A. Astrocytes and microglia: Models and tools. J. Exp. Med. 2019, 216, 71–83.
- Verkhratsky, A.; Nedergaard, M.; Hertz, L. Why are astrocytes important? Neurochem. Res. 2015, 40, 389–401.
- Prinz, M.; Jung, S.; Priller, J. Microglia biology: One century of evolving concepts. Cell 2019, 179, 292–311.
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318.
- Franze, K. The mechanical control of nervous system development. Development 2013, 140, 3069–3077.
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol. Rev. 2018, 99, 21–78.
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Pericyte-specific expression of PDGF beta receptor in mouse models with normal and deficient PDGF beta receptor signaling. Mol. Neurodegener. 2010, 5, 32.
- Alahmari, A. Blood-Brain Barrier Overview: Structural and Functional Correlation. Neural Plast 2021, 2021, 6564585.
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412.
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69.
- Kaya, M.; Ahishali, B. Basic physiology of the blood-brain barrier in health and disease: A brief overview. Tissue Barriers 2021, 9, 1840913.
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56.
- Chow, B.W.; Gu, C. The molecular constituents of the blood–brain barrier. Trends Neurosci. 2015, 38, 598–608.
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature 2020, 583, 425–430.
- Petersen, M.A.; Ryu, J.K.; Akassoglou, K. Fibrinogen in neurological diseases: Mechanisms, imaging and therapeutics. Nat. Rev. Neurosci. 2018, 19, 283–301.
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169.
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738.
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60.
- Vardy, E.R.; Kellett, K.A.; Cocklin, S.L.; Hooper, N.M. Alkaline phosphatase is increased in both brain and plasma in Alzheimer’s disease. Neurodegener. Dis. 2011, 9, 31–37.
- Stahr, N.; Galkina, E.V. Immune Response at the Crossroads of Atherosclerosis and Alzheimer’s Disease. Front. Cardiovasc. Med. 2022, 9, 870144.
- Iadecola, C. Atherosclerosis and neurodegeneration: Unexpected conspirators in Alzheimer’s dementia. Arterioscler Thromb Vasc Biol 2003, 23, 1951–1953.
- Dolan, H.; Crain, B.; Troncoso, J.; Resnick, S.M.; Zonderman, A.B.; Obrien, R.J. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of Aging cohort. Ann. Neurol. 2010, 68, 231–240.
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 653–666.
- de Bruijn, R.F.; Ikram, M.A. Cardiovascular risk factors and future risk of Alzheimer’s disease. BMC Med. 2014, 12, 130.
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604.
- van Sloten, T.T.; Sedaghat, S.; Carnethon, M.R.; Launer, L.J.; Stehouwer, C.D. Cerebral microvascular complications of type 2 diabetes: Stroke, cognitive dysfunction, and depression. Lancet Diabetes Endocrinol. 2020, 8, 325–336.
- Rhea, E.M.; Salameh, T.S.; Logsdon, A.F.; Hanson, A.J.; Erickson, M.A.; Banks, W.A. Blood-brain barriers in obesity. AAPS J. 2017, 19, 921–930.
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377.
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171.
- Santiago, J.A.; Potashkin, J.A. The Impact of Disease Comorbidities in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 631770.
- Rosenberg, G.A. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009, 8, 205–216.
- Candelario-Jalil, E.; Thompson, J.; Taheri, S.; Grossetete, M.; Adair, J.C.; Edmonds, E.; Prestopnik, J.; Wills, J.; Rosenberg, G.A. Matrix metalloproteinases are associated with increased blood–brain barrier opening in vascular cognitive impairment. Stroke 2011, 42, 1345–1350.
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72, 3–12.
- Zhao, N.; Liu, C.-C.; Qiao, W.; Bu, G. Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol. Psychiatry 2018, 83, 347–357.
- Alagarsamy, J.; Jaeschke, A.; Hui, D.Y. Apolipoprotein E in Cardiometabolic and Neurological Health and Diseases. Int. J. Mol. Sci. 2022, 23, 9892.
- Vecchio, F.L.; Bisceglia, P.; Imbimbo, B.P.; Lozupone, M.; Latino, R.R.; Resta, E.; Leone, M.; Solfrizzi, V.; Greco, A.; Daniele, A. Are apolipoprotein E fragments a promising new therapeutic target for Alzheimer’s disease? Ther. Adv. Chronic Dis. 2022, 13, 20406223221081605.
- Fernández-Calle, R.; Konings, S.C.; Frontiñán-Rubio, J.; García-Revilla, J.; Camprubí-Ferrer, L.; Svensson, M.; Martinson, I.; Boza-Serrano, A.; Venero, J.L.; Nielsen, H.M. APOE in the bullseye of neurodegenerative diseases: Impact of the APOE genotype in Alzheimer’s disease pathology and brain diseases. Mol. Neurodegener. 2022, 17, 62.
- Liu, C.-C.; Zhao, J.; Fu, Y.; Inoue, Y.; Ren, Y.; Chen, Y.; Doss, S.V.; Shue, F.; Jeevaratnam, S.; Bastea, L. Peripheral apoE4 enhances Alzheimer’s pathology and impairs cognition by compromising cerebrovascular function. Nat. Neurosci. 2022, 25, 1020–1033.
- Jackson, R.J.; Meltzer, J.C.; Nguyen, H.; Commins, C.; Bennett, R.E.; Hudry, E.; Hyman, B.T. APOE4 derived from astrocytes leads to blood-brain barrier impairment. Brain 2022, 145, 3582–3593.
- Greene, C.; Campbell, M. Tight junction modulation of the blood brain barrier: CNS delivery of small molecules. Tissue Barriers 2016, 4, e1138017.
- González-Mariscal, L.; Betanzos, A.; Nava, P.; Jaramillo, B.E. Tight junction proteins. Prog. Biophys. Mol. Biol. 2003, 81, 1–44.
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739.
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507.
- Ayka, A.; Şehirli, A. The Role of the SLC Transporters Protein in the Neurodegenerative Disorders. Clin. Psychopharmacol. Neurosci. Off. Sci. J. Korean Coll. Neuropsychopharmacol. 2020, 18, 174–187.
- Gil-Martins, E.; Barbosa, D.J.; Silva, V.; Remião, F.; Silva, R. Dysfunction of ABC transporters at the blood-brain barrier: Role in neurological disorders. Pharmacol. Ther. 2020, 213, 107554.
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood–brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345.
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9, S3.
- Al-Majdoub, Z.M.; Al Feteisi, H.; Achour, B.; Warwood, S.; Neuhoff, S.; Rostami-Hodjegan, A.; Barber, J. Proteomic Quantification of Human Blood–Brain Barrier SLC and ABC Transporters in Healthy Individuals and Dementia Patients. Mol. Pharm. 2019, 16, 1220–1233.
- Kook, S.-Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. Aβ1–42-RAGE Interaction Disrupts Tight Junctions of the Blood–Brain Barrier Via Ca2+-Calcineurin Signaling. J. Neurosci. 2012, 32, 8845–8854.
- Yamazaki, Y.; Shinohara, M.; Shinohara, M.; Yamazaki, A.; Murray, M.E.; Liesinger, A.M.; Heckman, M.G.; Lesser, E.R.; Parisi, J.E.; Petersen, R.C.; et al. Selective loss of cortical endothelial tight junction proteins during Alzheimer’s disease progression. Brain J. Neurol. 2019, 142, 1077–1092.
- Knox, E.G.; Aburto, M.R.; Clarke, G.; Cryan, J.F.; O’Driscoll, C.M. The blood-brain barrier in aging and neurodegeneration. Mol. Psychiatry 2022, 27, 2659–2673.
- Zhou, R.; Chen, L.L.; Yang, H.; Li, L.; Liu, J.; Chen, L.; Hong, W.J.; Wang, C.G.; Ma, J.J.; Huang, J.; et al. Effect of High Cholesterol Regulation of LRP1 and RAGE on Aβ Transport Across the Blood-Brain Barrier in Alzheimer’s Disease. Curr. Alzheimer Res. 2021, 18, 428–442.
- Boado, R.J. Molecular regulation of the blood-brain barrier GLUT1 glucose transporter by brain-derived factors. In Ageing and Dementia; Springer: Vienna, Austria, 1998; pp. 323–331.
- Chen, Y.; Joo, J.; Chu, J.M.; Chang, R.C.; Wong, G.T. Downregulation of the glucose transporter GLUT 1 in the cerebral microvasculature contributes to postoperative neurocognitive disorders in aged mice. J. Neuroinflammation 2023, 20, 237.
- Deane, R.; Sagare, A.; Zlokovic, B.V. The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 1601–1605.
- Boucher, P.; Herz, J. Signaling through LRP1: Protection from atherosclerosis and beyond. Biochem. Pharmacol. 2011, 81, 1–5.
- Storck, S.E.; Kurtyka, M.; Pietrzik, C.U. Brain endothelial LRP1 maintains blood–brain barrier integrity. Fluids Barriers CNS 2021, 18, 27.
- Sita, G.; Graziosi, A.; Hrelia, P.; Morroni, F. NLRP3 and Infections: β-Amyloid in Inflammasome beyond Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 6984.
- Merlini, M.; Rafalski, V.A.; Rios Coronado, P.E.; Gill, T.M.; Ellisman, M.; Muthukumar, G.; Subramanian, K.S.; Ryu, J.K.; Syme, C.A.; Davalos, D.; et al. Fibrinogen Induces Microglia-Mediated Spine Elimination and Cognitive Impairment in an Alzheimer’s Disease Model. Neuron 2019, 101, 1099–1108.e1096.
- Al-Ghraiybah, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 10572.
- Heithoff, B.P.; George, K.K.; Phares, A.N.; Zuidhoek, I.A.; Munoz-Ballester, C.; Robel, S. Astrocytes are necessary for blood–brain barrier maintenance in the adult mouse brain. Glia 2021, 69, 436–472.
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501.
- Liang, T.; Zhang, Y.; Wu, S.; Chen, Q.; Wang, L. The Role of NLRP3 Inflammasome in Alzheimer’s Disease and Potential Therapeutic Targets. Front. Pharmacol. 2022, 13, 845185.
- Al Rihani, S.B.; Darakjian, L.I.; Kaddoumi, A. Oleocanthal-Rich Extra-Virgin Olive Oil Restores the Blood-Brain Barrier Function through NLRP3 Inflammasome Inhibition Simultaneously with Autophagy Induction in TgSwDI Mice. ACS Chem. Neurosci. 2019, 10, 3543–3554.
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763.
- Wood, C.A.P.; Zhang, J.; Aydin, D.; Xu, Y.; Andreone, B.J.; Langen, U.H.; Dror, R.O.; Gu, C.; Feng, L. Structure and mechanism of blood-brain-barrier lipid transporter MFSD2A. Nature 2021, 596, 444–448.
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative Stress-Mediated Blood-Brain Barrier (BBB) Disruption in Neurological Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4356386.
- Alcendor, D.J. Interactions between Amyloid-Β Proteins and Human Brain Pericytes: Implications for the Pathobiology of Alzheimer’s Disease. J. Clin. Med. 2020, 9, 1490.
- Ding, R.; Hase, Y.; Ameen-Ali, K.E.; Ndung’u, M.; Stevenson, W.; Barsby, J.; Gourlay, R.; Akinyemi, T.; Akinyemi, R.; Uemura, M.T.; et al. Loss of capillary pericytes and the blood–brain barrier in white matter in poststroke and vascular dementias and Alzheimer’s disease. Brain Pathol. 2020, 30, 1087–1101.
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518.
- Blanchard, J.W.; Bula, M.; Davila-Velderrain, J.; Akay, L.A.; Zhu, L.; Frank, A.; Victor, M.B.; Bonner, J.M.; Mathys, H.; Lin, Y.-T.; et al. Reconstruction of the human blood–brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat. Med. 2020, 26, 952–963.
- Liu, L.; Wan, W.; Xia, S.; Kalionis, B.; Li, Y. Dysfunctional Wnt/β-catenin signaling contributes to blood–brain barrier breakdown in Alzheimer’s disease. Neurochem. Int. 2014, 75, 19–25.
- Wang, Q.; Huang, X.; Su, Y.; Yin, G.; Wang, S.; Yu, B.; Li, H.; Qi, J.; Chen, H.; Zeng, W.; et al. Activation of Wnt/β-catenin pathway mitigates blood–brain barrier dysfunction in Alzheimer’s disease. Brain J. Neurol. 2022, 145, 4474–4488.
- Gastfriend, B.D.; Nishihara, H.; Canfield, S.G.; Foreman, K.L.; Engelhardt, B.; Palecek, S.P.; Shusta, E.V. Wnt signaling mediates acquisition of blood–brain barrier properties in naïve endothelium derived from human pluripotent stem cells. eLife 2021, 10, e70992.
- Hussain, B.; Fang, C.; Huang, X.; Feng, Z.; Yao, Y.; Wang, Y.; Chang, J. Endothelial β-Catenin Deficiency Causes Blood-Brain Barrier Breakdown via Enhancing the Paracellular and Transcellular Permeability. Front. Mol. Neurosci. 2022, 15, 895429.
- Yang, A.C.; Vest, R.T.; Kern, F.; Lee, D.P.; Agam, M.; Maat, C.A.; Losada, P.M.; Chen, M.B.; Schaum, N.; Khoury, N. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 2022, 603, 885–892.
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; Winkler, E.A.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci. 2015, 18, 978–987.
- Juul Rasmussen, I.; Tybjærg-Hansen, A.; Rasmussen, K.L.; Nordestgaard, B.G.; Frikke-Schmidt, R. Blood-brain barrier transcytosis genes, risk of dementia and stroke: A prospective cohort study of 74,754 individuals. Eur. J. Epidemiol. 2019, 34, 579–590.
- O’Driscoll, M.C.; Daly, S.B.; Urquhart, J.E.; Black, G.C.; Pilz, D.T.; Brockmann, K.; McEntagart, M.; Abdel-Salam, G.; Zaki, M.; Wolf, N.I. Recessive mutations in the gene encoding the tight junction protein occludin cause band-like calcification with simplified gyration and polymicrogyria. Am. J. Hum. Genet. 2010, 87, 354–364.
- Wyss, L.; Schäfer, J.; Liebner, S.; Mittelbronn, M.; Deutsch, U.; Enzmann, G.; Adams, R.H.; Aurrand-Lions, M.; Plate, K.H.; Imhof, B.A. Junctional adhesion molecule (JAM)-C deficient C57BL/6 mice develop a severe hydrocephalus. PLoS ONE 2012, 7, e45619.
- Akawi, N.A.; Canpolat, F.E.; White, S.M.; Quilis-Esquerra, J.; Morales Sanchez, M.; Gamundi, M.J.; Mochida, G.H.; Walsh, C.A.; Ali, B.R.; Al-Gazali, L. Delineation of the clinical, molecular and cellular aspects of novel JAM 3 mutations underlying the autosomal recessive hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. Hum. Mutat. 2013, 34, 498–505.
- Chabriat, H.; Joutel, A.; Dichgans, M.; Tournier-Lasserve, E.; Bousser, M.-G. Cadasil. Lancet Neurol. 2009, 8, 643–653.
- Ghosh, M.; Balbi, M.; Hellal, F.; Dichgans, M.; Lindauer, U.; Plesnila, N. Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Ann. Neurol. 2015, 78, 887–900.
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127.
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116.
- Winfree, R.L.; Dumitrescu, L.; Blennow, K.; Zetterberg, H.; Gifford, K.A.; Pechman, K.R.; Jefferson, A.L.; Hohman, T.J. Biological correlates of elevated soluble TREM2 in cerebrospinal fluid. Neurobiol. Aging 2022, 118, 88–98.
- Wu, R.; Li, X.; Xu, P.; Huang, L.; Cheng, J.; Huang, X.; Jiang, J.; Wu, L.-J.; Tang, Y. TREM2 protects against cerebral ischemia/reperfusion injury. Mol. Brain 2017, 10, 20.
- Wang, Q.; Yang, W.; Zhang, J.; Zhao, Y.; Xu, Y. TREM2 overexpression attenuates cognitive deficits in experimental models of vascular dementia. Neural Plast. 2020, 2020.
- Taylor, X.; Cisternas, P.; You, Y.; You, Y.; Xiang, S.; Marambio, Y.; Zhang, J.; Vidal, R.; Lasagna-Reeves, C.A. A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J. Neuroinflam. 2020, 17, 223.
- Rauchmann, B.-S.; Sadlon, A.; Perneczky, R.; Initiative, A.s.D.N. Soluble TREM2 and inflammatory proteins in Alzheimer’s disease cerebrospinal fluid. J. Alzheimer’s Dis. 2020, 73, 1615–1626.
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276.
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 2023, 388, 9–21.
- Erickson, M.A.; Banks, W.A. Blood–brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513.

