The blood–brain barrier (BBB) is a unique and selective feature of the central nervous system’s vasculature. BBB dysfunction has been observed as an early sign of Alzheimer’s Disease (AD) before the onset of dementia or neurodegeneration. The intricate relationship between the BBB and the pathogenesis of AD, especially in the context of neurovascular coupling and the overlap of pathophysiology in neurodegenerative and cerebrovascular diseases, underscores the urgency to understand the BBB’s role more deeply. Preserving or restoring the BBB function emerges as a potentially promising strategy for mitigating the progression and severity of AD. Molecular and genetic changes, such as the isoform ε4 of apolipoprotein E (ApoEε4), a significant genetic risk factor and a promoter of the BBB dysfunction, have been shown to mediate the BBB disruption. Additionally, receptors and transporters like the low-density lipoprotein receptor-related protein 1 (LRP1), P-glycoprotein (P-gp), and the receptor for advanced glycation end products (RAGEs) have been implicated in AD’s pathogenesis.
- Alzheimer’s disease
- blood–brain barrier
- BBB
1. Introduction
2. NVU and the BBB
A schematic presentation of the NVU and the BBB are shown in Figure 1. The NVU is a crucial anatomical and functional unit that safeguards the homeostasis and optimal functioning of the central nervous system (CNS) [19][24]. Comprising several cell types, the NVU forms an interactive and dynamic system with its cellular constituents (Figure 1A). Neurons, the principal functional entities orchestrating signal transmission throughout the nervous system, are essential for the NVU [20][25]. Their close interaction with astrocytes, star-shaped glial cells, is pivotal for maintaining extracellular ion balance, facilitating synaptic transmission, and regulating CBF [21][26]. Microglia, the CNS’s resident immune cells, continually patrol the neural milieu and are ready to respond to alterations or threats [22][27]. Microglia participate in inflammatory responses and contribute to the NVU’s overall functionality by interacting with other cellular entities, including neurons and astrocytes [23][28].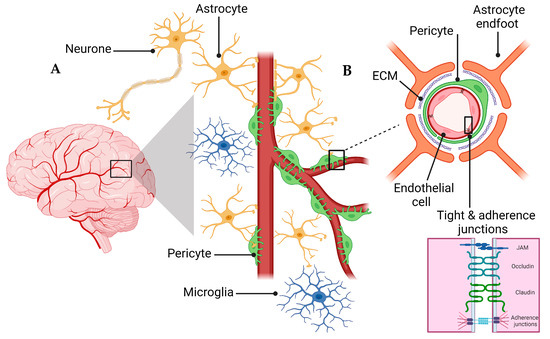
3. BBB Dysregulation with Aging
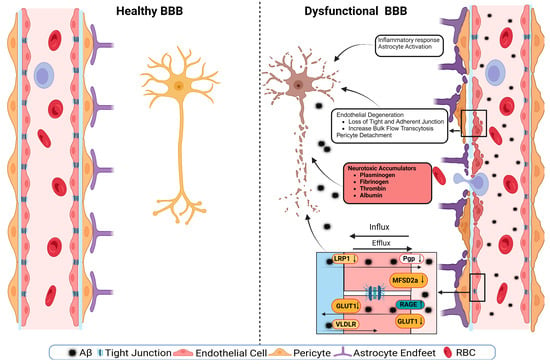
The dysregulation of BBB in aging involves both a loss of selective transport mechanisms and a reduction in structural integrity [33][44]. This is exemplified by a global shift in the pattern of protein transcytosis, transitioning away from a receptor-mediated transport (RMT) to an increased caveolar transcytosis, which allows the entry of potentially neurotoxic proteins such as albumin and fibrinogen that are otherwise restricted in youth [33][34][44,45]. This change in the BBB’s permeability is implicated in the pathophysiology of neurodegenerative diseases, including AD, by facilitating neuroinflammation through unregulated protein entry [35][36][46,47]. Moreover, the age-related decrease in pericyte coverage further compromises the BBB, impairing blood flow and neuronal function [37][48]. The observed upregulation of endothelial alkaline phosphatase, namely ALPL, in AD patients, suggests it to be a potential therapeutic target, as its inhibition could modify the BBB’s permeability and influence the disease progression [38][49]. Overall, these changes underscore a “two-hit” hypothesis for vascular contributions to AD, where both pericyte loss and dysfunctional transcytosis act synergistically with other pathogenic factors like Aβ accumulation. Understanding the evolving nature of the BBB’s communication with the periphery is crucial for deciphering the impact of aging on the brain in health and disease [38][49].4. BBB Dysfunction in AD
4.1. Effect of Comorbidities on BBB
The BBB disruption is related to multiple comorbidities, including vascular comorbidities such as atherosclerosis, where both diseases are characterized by inflammation and vascular dysfunction [39][58]. Small vessel dysfunction is responsible for the BBB breakdown, CBF and Aβ clearance reduction, and neuronal dysfunction [39][40][58,59]. However, findings from the Baltimore Longitudinal Study of Aging (BLSA) Cohort, which investigated the relationship between Alzheimer’s and atherosclerosis, found no association between the degree of Alzheimer’s pathology and atherosclerosis in the aorta, heart, and intracranial vessels, though only intracranial atherosclerosis correlated with dementia [41][60]. Moreover, comorbidities, including Type 2 Diabetes Mellitus (T2DM), hypertension, obesity, sleep disorders, and hypercholesterolemia, are increasingly recognized as contributing factors in the disruption of the BBB and the progression of AD [42][43][61,62]. In the case of T2DM, which is characterized by insulin resistance and chronic hyperglycemia, it causes endothelial dysfunction and inflammation that could ultimately compromise the BBB’s integrity. These alterations potentially facilitate the entry of neurotoxic molecules like Aβ, thereby contributing to AD pathology [44][45][63,64]. Obesity, especially as a part of the metabolic syndrome, often precipitates systemic inflammation and endothelial dysfunction, contributing to a compromised BBB function [46][66]. Sleep disorders, particularly those impacting the glymphatic system, are emerging as critical players in hindering the efficient clearance of neurotoxic products such as Aβ, ultimately fostering an environment conducive to AD progression [47][67]. The cumulative impact of these comorbidities cannot be overlooked, as they often co-occur and may have synergistic effects on BBB dysfunction and AD pathology. These comorbidities exacerbate the degradation of TJs, thereby compounding the risk of BBB dysfunction and the subsequent development and progression of AD [48][49][50][51][69,70,71,72]. The interplay between the degradation of TJs by proteases and the risk factors for AD suggests a vicious cycle where the compromised barrier function of the BBB can lead to further neuronal damage and progression of AD. At the same time, the disease state promotes a further BBB breakdown [51][72].4.2. ApoE and BBB Dysfunction in AD
ApoE is mainly expressed in astrocytes and microglia in the brain; its functions are associated with the endocytosis of lipoproteins, the deposition and transport of Aβ, membrane integrity, neurotoxicity, and synaptic plasticity [14][19]. It is produced in the brain, liver, kidneys, skin, adipose tissue, and many other organs [52][74]. Within the CNS, ApoE is produced de novo separately and independently from peripheral ApoE [53][75]. ApoE protein has three different isoforms: ApoEε2, ApoEε3, and ApoEε4. These isoforms have functional and structural differences, inconsistencies, and discrepancies in their interaction with low-density lipoprotein (LDL) receptors [52][74]. Within the cell, ApoE plays a role in cellular processes involving the maintenance of the cytoskeleton, mitochondria, and dendrites, leading it to play a prominent role in the overall health of neurons [52][74]. ApoE amino acids vary at locations 112 and 158 on chromosome 19, and there are six different genotypes, i.e., three homozygous and three heterozygous [54][76]. ApoEε2 is protective against the pathology of AD [55][77]. ApoEε2 expression clears Aβ normally and may suppress inflammation, which can protect against AD [54][76]. To investigate the role of ApoEε2 in AD, using post-mortem human cortices, Fernández-Calle, and colleagues reported that the abundance of ApoEε2 and its ability to bind to LDLRs increased the efficiency of Aβ clearance. In addition, they found that the expression of mRNA molecules related to ECM increased, which could contribute to the protection of the BBB’s integrity. These effects of ApoEε2 point toward its protective abilities against AD [56][79]. ApoEε3, the most common form of the gene, on the other hand, does not present a risk for or protection from AD relative to the ε4 and ε2 alleles, respectively. ApoEε3 protein functions well in binding to LDL, very low-density lipoprotein (VLDL), and low-density lipoprotein-related protein-1 (LRP1) receptors, which play a role in clearing Aβ [56][79]. The most heavily researched isoform is ApoEε4. ApoEε4 is associated with an earlier onset of AD due to a higher plaque density earlier in life [14][19]. ApoEε4 likely contributes to AD pathology by increasing neurotoxicity and decreasing the protection of the CNS [53][75]. However, the mouse apoE gene exists only in one isoform and is located on chromosome 7 rather than 19. For this reason, mice are often artificially introduced to human ApoE to make the study applicable to human AD [56][79]. ApoEε4 correlates with a decreased BBB integrity and cognitive impairment due to disturbed homeostasis and neurotoxin extravasation into the brain [57][80]. ApoEε4 knock-in mice displayed the loss of pericytes and a subsequent BBB degradation as they aged [58][81]. This loss of pericytes was also found in human ApoEε4 carriers [53][75].4.3. Tight Junctions’ Role in BBB Health and Disease
The barrier function of TJs in the BBB is multifaceted. They restrict the intercellular space, preventing the unregulated diffusion of substances between the blood and the brain. This role is critical in preserving the specialized environment required for an optimal neural function [59][84]. Concurrently, TJs maintain cellular polarity by ensuring an asymmetrical distribution of proteins across the cell, a decisive factor for a directional transport across the endothelium [59][84]. On the paracellular front, TJs exert tight control over the diffusion pathways by leveraging an array of integral proteins, such as claudins, occludin, and JAMs. The specific assembly of these proteins determines the permeability properties of the junctions, allowing selective passage of ions and molecules while safeguarding against harmful substances [60][85]. For the transcellular route, the asymmetrical distribution of transporters and channels underpins a regulated transport mechanism, ensuring that essential nutrients reach the brain while metabolic waste products are efficiently evacuated. The concerted action of these transcellular elements with TJs establishes the BBB as a dynamic regulatory interface, which is adept at adjusting to physiological needs and protecting against pathological insults. In AD, one of the critical pathways by which TJs can be compromised involves the activity of matrix metalloproteinases (MMPs) and other proteases [61][86]. MMPs are a family of zinc-dependent endopeptidases capable of degrading various components of the extracellular matrix (ECM). They are known to modulate the permeability of the BBB by targeting TJ proteins [62][87]. MMPs, particularly MMP2 and MMP9, have been demonstrated to cleave occludin, claudins, and ZO proteins, leading to the disassembly of TJs and an increased BBB permeability [62][87]. The activity of MMPs is tightly regulated under normal physiological conditions but can be upregulated in response to inflammation, ischemia, and oxidative stress, which are common in the AD pathology [61][86].4.4. Transporters’ Role in BBB Health and Disease
The functionality of the BBB is partly enabled by the presence of diverse influx and efflux transporters expressed in the endothelial cells, notably from the solute carrier (SLC) and ATP-binding cassette (ABC) superfamilies [63][64][65][91,92,93]. The BBB restricts drug access to the brain by permitting only lipophilic molecules of low molecular weight into the brain via the transcellular pathway from the bloodstream, where most researchers consider a cut-off point of 400–600 kDa. However, deviations from this cut-off point have been published [66][94]. Small, lipid-soluble drugs, such as antidepressants, penetrate the BBB via passive diffusion across the BBB [31][38]. Transcytosis transports macromolecules, such as insulin and amino acids [31][38]. Efflux transporters counteract the passive diffusion by forcing foreign substances, toxic metabolites, and other waste products out of the brain [31][38]. In contrast, recombinant proteins, therapeutic antibodies, and nucleic acid drugs are too large to cross the BBB [31][38]. The BBB disruption could be due to, at least in part, the modulation of transporters’ function or expression, accumulating waste products, and the inadequate nutrient delivery to the brain [31][38]. Overall, more than 50 transporters are expressed in the BBB, and their modulation by aging or AD could alter the BBB function, thus contributing to AD pathology [67][95].5. BBB Breakdown Mechanisms
A schematic presentation of the discussed mechanisms is presented in Figure 2. The disruption of the BBB could impair any of the NVU cellular components. The disruption of endothelial cells might result in a decreased TJs’ expression. It has been shown that occludin, claudin-5, and ZO1 expressions are lower in the Aβ-laden capillaries of patients with capillary CAA [31][38], implying that reduced TJs may increase the vascular permeability in an AD brain. Multiple cellular signaling pathways, such as calcium signaling, could mediate the disruption of TJs. In AD, the RAGE–Aβ42 interaction disrupts TJs via a calcium-calcineurin signaling pathway [68][133]. In addition, in the monolayer culture of bEnd3 cells, adding Aβ42 induces structural alterations in ZO1 [68][133]. Other TJ proteins, such as claudin-5 and occludin, were also structurally altered, and their expression was reduced by Aβ42 [68][133]. This result was confirmed as neutralizing antibodies against RAGEs and calcineurin and MMP inhibitors prevented Aβ42-induced changes in the ZO1 expression [68][133]. Furthermore, the expression of TJs is negatively correlated with Aβ40 levels in cortical areas and positively correlated with synaptic markers in patients with AD, highlighting the role of TJs in the AD pathology development [69][134].