Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kawther Sayed Zaher | -- | 3049 | 2023-12-12 00:46:04 | | | |
| 2 | Rita Xu | Meta information modification | 3049 | 2023-12-12 02:42:04 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zaher, K.; Basingab, F. Gut Microbiota and Dendritic Cells in Colorectal Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/52596 (accessed on 23 July 2026).
Zaher K, Basingab F. Gut Microbiota and Dendritic Cells in Colorectal Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/52596. Accessed July 23, 2026.
Zaher, Kawther, Fatemah Basingab. "Gut Microbiota and Dendritic Cells in Colorectal Cancer" Encyclopedia, https://encyclopedia.pub/entry/52596 (accessed July 23, 2026).
Zaher, K., & Basingab, F. (2023, December 12). Gut Microbiota and Dendritic Cells in Colorectal Cancer. In Encyclopedia. https://encyclopedia.pub/entry/52596
Zaher, Kawther and Fatemah Basingab. "Gut Microbiota and Dendritic Cells in Colorectal Cancer." Encyclopedia. Web. 12 December, 2023.
Copy Citation
Colorectal cancer (CRC) is a malignancy that manifests in serial stages and has been observed to have an escalating incidence in modern societies, causing a significant global health problem. The development of CRC is influenced by various exogenous factors, including lifestyle, diet, nutrition, environment, and microbiota, that can affect host cells, including immune cells.
C-type lectin receptor
CRC
DCs
interleukins
1. Introduction
Colorectal cancer (CRC) significantly contributes to cancer-related deaths and ranks as the third most prevalent cancer globally [1]. CRC is notably high among populations that adopt a Western lifestyle; it is also rising in other locations, primarily low-income nations, creating a worldwide health threat [2]. CRC develops slowly over years, as adenomas originate from tiny, hard-to-see neoplastic foci into malignant carcinomas that may spread throughout the body [3]. CRC is reported to have considerable heterogeneity due to genetic instability [4][5]. It may also be caused by lifestyle, food, nutritional intake, environmental circumstances, and the microbiome. These variables affect non-neoplastic cells, including immune cells, increasing heterogeneity [6][7]. CRC is caused mainly by immune system malfunction, as it begins by impairing the host’s anti-tumor immunity, the so-called tumor microenvironment [8]. The tumor microenvironment changes early in neoplastic transformation and progression to promote cell proliferation. Tumor development and metastasis or immune-mediated cancer inhibition may follow. Inflammatory and immunological cells in the tumor microenvironment may accelerate colorectal cancer. These cells may limit tumor development or cause chronic inflammation, suppressing the immune system and promoting CRC progression [9][10].
2. Causes and Symptoms of CRC
Diet and lifestyle have been associated with CRC risk for decades. Modifiable risk factors cause 50–60% of CRC cases [11]. Tobacco use, excessive alcohol use, obesity, lack of exercise, consuming red and processed meat, and insufficient dietary fiber and other nutrients worsen health (Figure 1). The microbiome, such as bacteria, viruses, and fungi, is essential to health. Microbiota disturbances may cause CRC. According to Song and Chan [12], environmental risk factors may cause and promote CRC by altering the gut microbiota. Personal or family history of colorectal polyps or CRC, Lynch syndrome, inflammatory bowel disease (IBD), racial/ethnic origin, and type 2 diabetes are all unchangeable risk factors for CRC [13]. CRC can develop in persons as young as in their 20s, if they are predisposed to the disease. Advanced age is the most prominent risk factor [14]. Over 90% of new cases and 94% of CRC-related fatalities occur in people over 50. Medium-risk people over 50 should be examined for CRC, and high-risk people should be monitored. High-risk factors include CRC syndromes, adenomatous polyps, IBD, and personal or family history of CRC [15]. Constipation, diarrhea, stomach pain, and rectal pain precede 70–95% of early-onset CRC. Later, anemia, rectal bleeding, and weight loss occur [16].
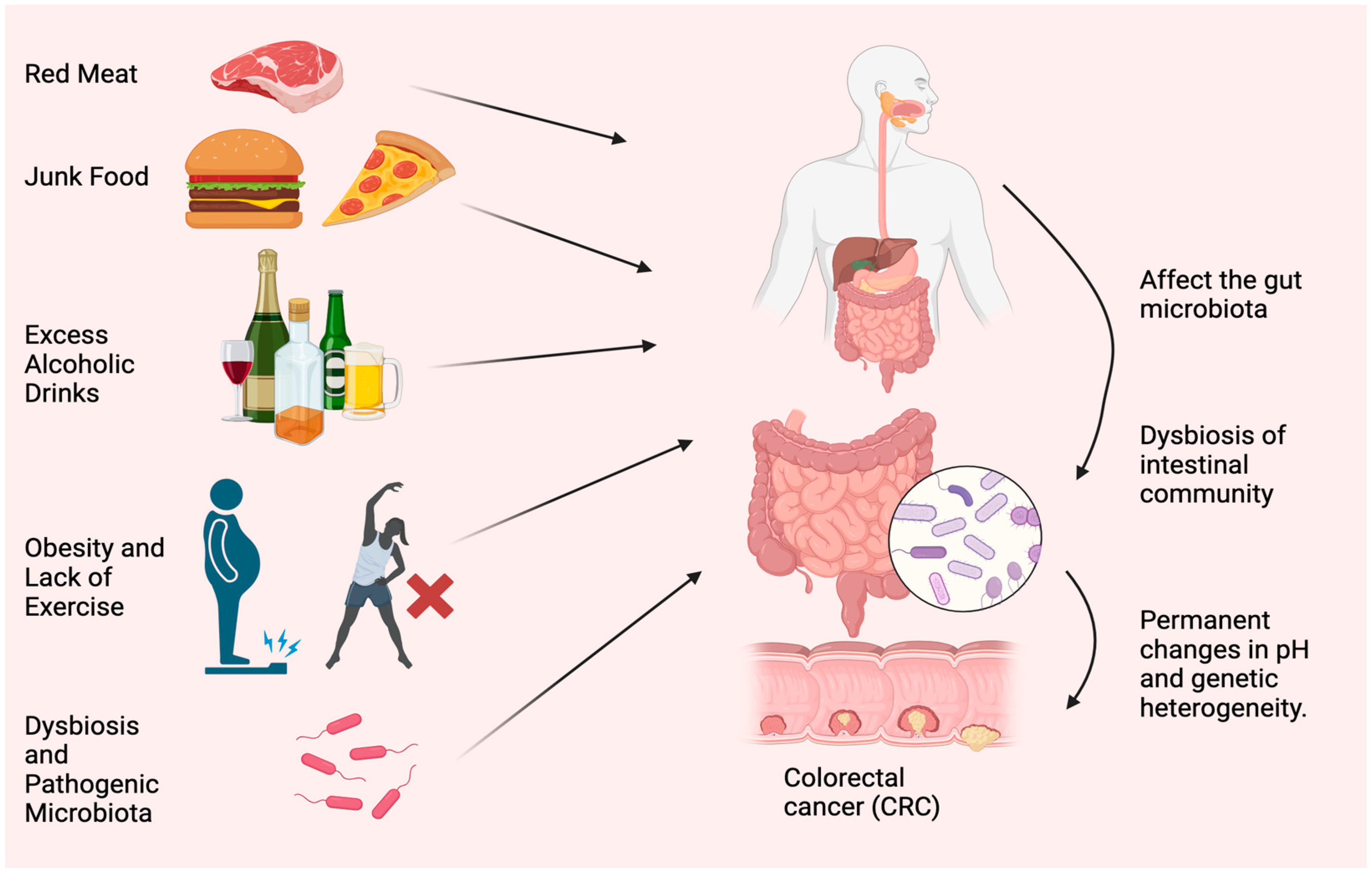
Figure 1. Most typical causes of CRC. Red meat, junk food, excess alcoholic drinks, obesity, lack of exercise, dysbiosis, and accumulation of pathogenic microorganisms are the main reasons for CRC.
2.1. Effect of Red Meat and Junk Food
Research by the World Cancer Committee [17] demonstrated that the regular consumption of 100 g of red and processed meat heightens the likelihood of developing CRC by 12%. This heightened susceptibility is attributable to the presence of heme iron, sulfur, and choline, in addition to nitrates, nitrites, emulsifiers, and heterocyclic amines and polycyclic aromatic hydrocarbons, which emanate from processing and cooking techniques and may stimulate the onset of cancer [18]. These meals may fuel gut microbes that generate carcinogens. Red meat provides choline and carnitine. Red meat elevated blood and urine trimethylamine-N-oxide (TMAO) in a random controlled experiment. TMAO increases heart disease and mortality. Two bacterial genes, cutC (choline TMA-lyase) and cutD (choline TMA-lyase-activating enzyme), were discovered in extremely high numbers, supporting the concept that the choline-TMAO pathway may modify metagenomes and cause CRC. Hungatella hathaway, Clostridium asparagiforme, Klebsiella oxytoca, and E. coli increased in CRC patients with sequence changes in these two genes. Several prospective studies have linked high dietary and blood choline levels to CRC [19]. DNA methylation and synthesis require choline, folate, and vitamin B12. TMAO only increased CRC risk in vitamin B12-deficient people. Its effects may vary, depending on whether it is in the carbon metabolism cycle or in the stomach for microbial TMAO synthesis [20]. CRC is connected to bacterial secondary bile acids, processed foods, red meat, and free radicals [21]. Bile acid content and concentration variations in metabolic diseases and infectious bowel disease (IBD) enhance CRC risk. Several analyses of eight geographically and methodologically diverse fecal shotgun genomic studies found that higher secondary bile acid was produced [22]. According to a study, high-fat diets may contribute to the epidemic of CRC in young people. It has been found that high-fat diets alter the gut bacteria and alter the digestive substances, called bile acids, in mice. Consequently, inflammation occurs, increasing the chance of developing colorectal cancer, a disease that is notoriously difficult to treat [23]. When the body consumes fat, bile acids are synthesized in the liver, and they play a role in facilitating lipid absorption by the small intestine. Bile acids are reabsorbed and subjected to enterohepatic circulation in the small intestine. Bile acids undergo complex biotransformation in the colon by gut bacteria, resulting in secondary bile acids that promote tumor growth. Excessive dietary fat intake leads to a high level of secondary bile acids in feces and primes the gut microbiota to produce bile acids. It is believed that this promotes an altered overall bile acid pool, resulting in an altered intestinal and hepatic cross-signaling of the farnesoid X receptor (FXR), the receptor for bile acids. The FXR gene is a main regulator of bile acid-mediated effects on intestinal tumorigenesis, integrating dietary, microbial, and genetic risk factors. In healthy and tumorigenic conditions in the intestine, selective FXR agonist or antagonist activity is determined by additional factors such as bile acid concentration, composition, and genetic instability of the cells [24].
2.2. Correlation between Healthy Diet and CRC
Many epidemiological inquiries remain to be conducted to substantiate this concept. Based on a meta-analysis of 21 studies, there appears to be no link between fiber consumption and CRC risk. Nevertheless, the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort has persistently demonstrated that a higher fiber intake can lower susceptibility to CRC [25]. Europeans eat cereals, but Americans eat fruits and vegetables, which may explain the observed behavior. US cohorts stated that low fiber consumption might have prevented efficacy. According to numerous studies, whole grains reduce the risk of CRC. A meta-analysis found that increasing a whole grain diet by 90 g/day reduced CRC risk by 17% [17].
2.3. Role of Obesity and Physical Activity on CRC
A meta-analysis revealed that a rise in body mass index (BMI) of 5 kg/m2 raised the risk of CRC by 5%. Additionally, obesity may potentially escalate the incidence of early-onset CRC [26]. Obesity is linked to inflammatory and metabolic changes in adipokines, insulin-like growth factor 1 signaling, systemic inflammation, sex hormones, and CRC risk. Obesity-induced gut microbiota alterations and metabolic byproducts may affect cancer development. Obesity decreases gut microbiota diversity [27].
Obesity-induced intestinal barrier dysfunction may worsen systemic inflammation. Leaking microbiota products such as lipopolysaccharide (LPS) causes metabolic endotoxemia. Greater BMIs are related to greater LPS-binding protein (LBP) and LPS levels. Losing weight decreases blood LBP and LPS [28]. Adenomas were associated with increased LPS levels in cross-sectional research. Villous adenomas had higher LPS levels than tubular ones. An LBP gene polymorphism (rs2232596) has been associated with CRC [29].
According to cohort data, high exercisers decrease CRC risk by 19% compared with non-exercisers. Exercise has not been linked to rectal cancer. In many cross-sectional studies, exercise influences the gut microbiome’s composition and function [17].
A 6-week intervention of endurance-based exercise was provided to a population of 32 people who lived sedentary lifestyles, after 2 weeks of the baseline study. The subjects then had a 6-week exercise-free washout period. The study found that only skinny participants had short-chain fatty acid concentrations in their stools after exercise. The number of butyrate-producing bacterial taxa increased, linked to body composition changes in lean people [30].
2.4. The Role of GUT Microbiota in Dysbiosis and CRC
Specific nutritional components can regulate the gut microbiome and promote a persistent inflammatory state by modulating immune and inflammatory pathways, making diet a significant factor in CRC onset, progression, and prevention. Human microbiota includes bacteria in the gastrointestinal, genitourinary, oral, respiratory, and cutaneous systems, among others. These bacteria interact with the body in various mechanisms and are essential for several physiological routes, including immunology, tissue growth, nutrition absorption, metabolism, and cancer [31]. Recent research showed that nutritional, genetic, and environmental factors affect the microbiome. Gut microorganisms also affect food metabolism. Nutrition affects microbiota in populations with various diets. According to the study, “healthy” diets like the Mediterranean diet increase microbial biodiversity. Dietary fiber, polyphenols, and lipids are crucial food–gut microbiome interactions. Different dietary lipids may affect gut microbiota variety, microorganisms, and activity. Metabolic effects may modulate systemic low-grade inflammation. Many pathways link dysbiosis to illnesses, including cancer [32]. Oncogenesis may also result from microbiota-induced mucosal inflammation or systemic metabolic and immunological disruption. The microbiome may indirectly alter cancer treatment and immunity. The inhibition of programmed cell death (PD-1) is associated with antitumor effects of epithelial malignancies in hepatocellular carcinoma and is also linked to Akkermansia muciniphila frequency [33]. Lower levels of this bacterium in rats and humans are connected to obesity, insulin resistance, type 2 diabetes, and other cardiometabolic diseases. CRC patients’ fecal and mucosal microbiota change during the disease, reducing bacterial diversity [34].
It is important to note that LPS is an important component of the outer layer of bacteria and has a strong pathogenic effect [35]. Inflammation and immune responses in the hosts can be triggered through TLR activation (one of the pattern recognition receptors), among which TLR4 and TLR2 are the most important receptors [36]. The FimA and Mfa1 subunits of bacterial fimbriae are important in promoting bacterial adhesion to host tissues and their invasion [37]. A TLR on endothelial cells [38], macrophages [39], and DCs [40] can also recognize it, activating the cells and causing them to produce cytokines and adhesion molecules. These cytokines stimulate the proliferation and differentiation of DCs. A link between innate and adaptive immunity is established when inflamed endothelial cells trigger macrophages and immature tissue DCs to present the bacterial cell wall on their surface for T cells called major histocompatibility molecules (MHC-I). DCs recognize pathogen-associated molecular patterns (PAMP) and damage-associated molecular patterns (DAMP) [41]. As DCs mature, metabolic, cellular, and gene transcription programs are activated, allowing them to migrate from peripheral tissues to secondary lymphoid organs, where T lymphocyte-activating antigen presentation may occur in response to CCL19 and CCL21 [42]. The maturation of DCs is characterized by the loss of adhesive structures, the reorganization of the cytoskeleton, and an increase in motility. As a result of DC maturation, the level of endocytic activity declines; however, the expression of MHC-II and co-stimulatory molecules such as CD80 and CD86, as well as the chemokine receptor CCR7, is increased, as are the levels of pro-inflammatory cytokines such as TNF-α and IL-12 [43]. A mature DC expresses higher levels of the chemokine receptor, CCR7 [44], and secretes cytokines required for T-cell activation [45]. Consequently, mature DCs interact with antigen-specific T cells to trigger antigen-specific immune responses [46]. By interacting with CD4+ T cells (called MHC-II), DCs may induce differentiation into different T helper (Th) cells [47], Th2 cells [48], Th17 cells [49], or other CD4+ T cells [50]. This way, DCs can trigger responses against intracellular antigens from various cell types [51] and even prime CD8+ lymphocytes without CD4+ T cells [52]. Cross-presentation also introduces tolerance to intracellular self-antigens not expressed by APC, called cross-tolerance [53]. The “immature” cells are, however, extremely effective at capturing antigens due to their high endocytic capacity via receptor-mediated endocytosis, including lectins [54], Toll-like receptors [55], FC receptors, and complement receptors [56]. Thus, immature DCs act as sentinels against invading pathogens and as tissue scavengers, capturing apoptotic and necrotic cells [57]. Accordingly, immature DCs are essential for inducing and maintaining immune tolerance [58]. DCs internalize cell apoptosis and the loss of critical details in polyps, such as tissue turnover. However, they do not induce DCs maturation [59]. In this way, their antigens are presented to T cells without the activating co-stimulatory signals delivered by a mature DC, resulting in the apoptosis of T cells and the development of regulatory T cells. It has been demonstrated that a “tolerogenic DC” expresses fewer co-stimulatory molecules and proinflammatory cytokines. Still, it upregulates the expression of inhibitory molecules (such as PD-L1 and CTLA-4) and secretes anti-inflammatory cytokines (IL-10, tumor growth factor beta, for example). In this situation, the DC fails to increase the co-stimulatory molecules required to activate T cells, meaning that the immune system leaves uncontrolled disease despite recognizing pathogens or changes in the tumor microenvironment [52][58].
2.4.1. Fusobacterium nucleatum
Cohort studies showed that colorectal neoplasia patients have more Fusobacterium nucleatum (F. nucleatum) in their feces than controls. This was connected to more advanced disease, less T-cell infiltration, a higher risk of recurrence, and a worse prognosis. This was linked to serrated pathway molecular characteristics. The inhibition of Myeloid-derived suppressor cells and natural killer (NK) and T cells enhances tumor growth. Modifying E-cadherin/-catenin may affect this mechanism [35]. According to recent studies, F. nucleatum stimulates the production of reactive oxygen species (ROS) and inflammatory cytokines (e.g., IL-6 and TNF) in colorectal cancer [38][60]. MLH1 is epigenetically silenced due to inflammation and ROS, reducing mismatch repair protein’s enzymatic activity (MMR) activity. In addition, virulence factors derived from F. nucleatum also inhibit T cell proliferation [33][61]. This may be consistent with a recent study finding that a greater abundance of F. nucleatum in colorectal carcinoma tissue was associated with a lower density of T cells in the tumor microenvironment. Based on these findings, F. nucleatum may downregulate antitumor T-cell-mediated adaptive immunity [62].
2.4.2. Enterotoxigenic Bacteroides fragilis (ETBF)
Among the symbiotic bacteria in the intestinal tract is Bacteroides fragilis. The majority of bacteria in the human body aid in the digestion of food and maintain intestinal health. Occasionally, however, these bacteria produce a toxin that disrupts the cells on the surface of the gut, resulting in the development of CRC. B fragilis has two subtypes, nontoxic B fragilis and enterotoxigenic B. fragilis (ETBF), which produce B. fragilis toxin (BFT). ETBFs produce a 20 kDa metalloprotease, B. fragilis toxin (BFT), which disturbs the intestinal environment and promotes inflammation [63]. BFT destroys the epithelial barrier and E-cadherin cleavage. Additionally, cleavage of E-cadherin can activate the Wnt signal transduction pathway, cause mucosal inflammation, and promote colon cancer development. Further, the STAT3 pathway is required to develop Th17 cells and colon transformation. Th17 cells are stimulated to produce IL-17 after ETBF stimulates the STAT3 pathway, which activates the NF-κB and Wnt pathways, creating intestinal inflammatory tumor microenvironments. Through its rapid activation of the STAT3 pathway, ETBF is involved in the production of IL-17 by Th17 cells. Injection of an anti-IL-17 antibody in mice can inhibit tumor formation by inhibiting IL-17 production [64]. ETBF can also upregulate the expression of spermine oxidase (SMO) in colonic epithelial cells, thus increasing the generation of SMO-dependent reactive oxygen species (ROS), causing DNA damage and, ultimately, leading to CRC development [65].
2.4.3. Escherichia coli (E. coli)
IBD and CRC patients have higher mucosa-associated E. coli levels. In CRC, E. coli invades the intestinal wall and becomes intracellular. Polyketide synthase gene complex (pks) bacteria generate genotoxin colibactin, more commonly in CRC patients. Later-stage tumors have more pks+ E. coli strains. Ki-67 expression correlates with internalized and mucosa-associated E. coli. One fecal microbiome investigation found E. coli enrichment in CRC patients. E. coli prefers to colonize the mucosal lining and, intracellularly, occupy intestinal cells rather than the lumen, impeding feces removal [66].
3. Pathogenesis and Genetic Alteration of CRC
CRC originates in pre-cancerous polyps. The term “polyp” refers to localized outgrowths or clusters of aberrant cells that extend into the intestinal cavity from the gut’s mucosal layer. CRC exhibits either sessile or pedunculated polyps [67]. If these polyps have enough genetic alterations, their multiplying cells may be able to permeate the intestinal wall, a characteristic of colorectal cancer. These cells may then alter, move to nearby lymph nodes, and reach distant metastatic locations. When the polyp grows, it may acquire genetic mutations and epigenetic alterations that cause cytologic and histologic dysplasia [68]. Slow cellular DNA damage may cause high-grade dysplasia, which increases the risk of invasive cancer [69]. Without removal, these polyps can invade the colon and rectal wall and spread. New blood arteries allow cancer cells to access the lymphatic and circulatory systems and metastasize to distant organs. Pre-cancerous polyps must be removed quickly to break the adenoma-carcinoma cycle and prevent colorectal cancer. Histologically, genetic and epigenetic alterations cause a polyp to become a malignancy (Figure 2). DNA alteration can be either inherited or acquired. Inherited mutations, including APC (adenomatous polyposis coli), PMS2, MSH2, MLH1and gene changes, cause 5% of CRC cases. Hereditary mutations with spontaneous APC and DNA divergence repair metamorphoses have illuminated the genetic path from premalignant polyps to cancer [70]. Sessile serrated polyps (SSPs) and adenomas polyps often cause CRC. The chromosomal instability route affects 65–70% of sporadic malignancies, which are often linked to conventional adenomas. Many mutations distinguish this method. Two genes cause CRC APC gene mutations to alter chromosomal segregation during cell division, while KRAS oncogene changes affect cellular proliferation, differentiation, motility, and survival. Mutations may affect transcription and apoptotic regulator p53. This may affect cellular processes and cause cancer [71]. However, BRAF gene mutations that disrupt growth signals and impede apoptosis typically cause sessile serrated polyps (SSPs). SSPs have KRAS mutations at 23, 21, 24, and 13, but less than adenomatous polyps (Figure 2). Serrated lesions cause gene promoter hypermethylation in CRC [72]. Methylation of the promoter region may inhibit gene transcription and function. Inactivating this gene affects many genes, including development-promoting genes [73]. CpG island methylators contain abnormally methylated genes such as BMP3 and NDRG4 [74]. Microsatellite instability (MSI) disrupts DNA repair genes, promoting genetic diversity in CRC. MSI may cause short, non-coding, repetitive DNA sequences to replicate unevenly, making bodies more sensitive to genetic alterations [75]. MSI is a phenomenon that can manifest in adenomatous and serrated polyps (Figure 2). It is closely associated with alterations in DNA mismatch repair genes resulting from either germline mutations, such as those observed in hereditary nonpolyposis colorectal cancer, or sporadic mutations resulting from abnormal methylation of the MLH1 promoter region. The latter is closely linked with the CpG island methylator phenotype [76].
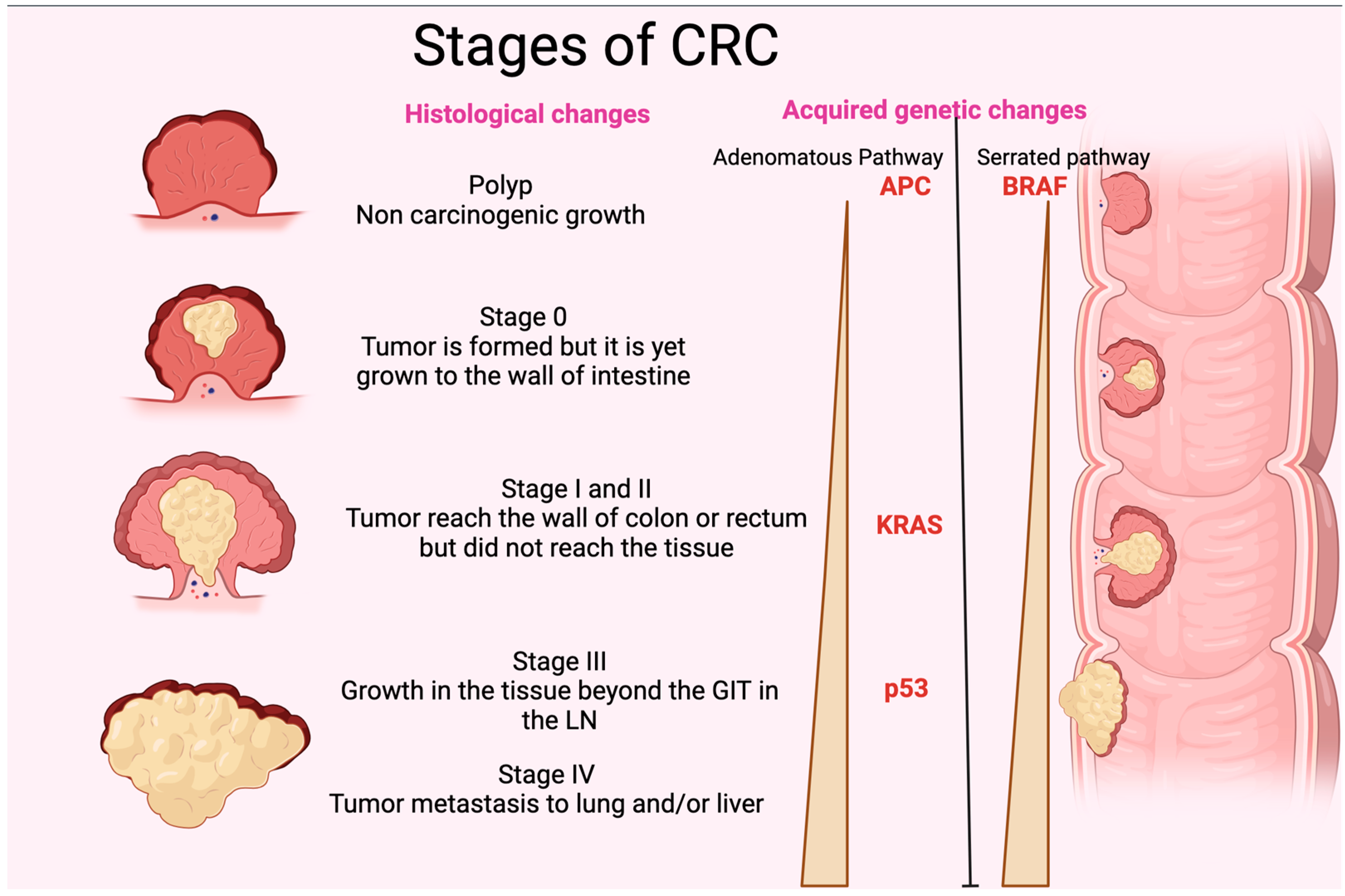
Figure 2. Stages of CRC. The figure shows the different histological stages of CRC. It also represents the gradual increase in p35, KRAS, and APC (adenomatous polyposis coli) in the adenomatous pathway and BRAF in the serrated pathway of acquired genetic changes. GIT: gastrointestinal tract; LN: lymph node.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- van der Geest, L.G.; Lam-Boer, J.; Koopman, M.; Verhoef, C.; Elferink, M.A.; de Wilt, J.H. Nationwide trends in incidence, treatment and survival of colorectal cancer patients with synchronous metastases. Clin. Exp. Metastasis 2015, 32, 457–465.
- Pandurangan, A.K.; Divya, T.; Kumar, K.; Dineshbabu, V.; Velavan, B.; Sudhandiran, G. Colorectal carcinogenesis: Insights into the cell death and signal transduction pathways: A review. World J. Gastrointest. Oncol. 2018, 10, 244–259.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558.
- Kong, C.; Liang, L.; Liu, G.; Du, L.; Yang, Y.; Liu, J.; Shi, D.; Li, X.; Ma, Y. Integrated metagenomic and metabolomic analysis reveals distinct gut-microbiome-derived phenotypes in early-onset colorectal cancer. Gut 2023, 72, 1129–1142.
- Kosumi, K.; Mima, K.; Baba, H.; Ogino, S. Dysbiosis of the gut microbiota and colorectal cancer: The key target of molecular pathological epidemiology. J. Lab. Precis. Med. 2018, 3, 76.
- Russo, E.; Gloria, L.D.; Nannini, G.; Meoni, G.; Niccolai, E.; Ringressi, M.N.; Baldi, S.; Fani, R.; Tenori, L.; Taddei, A.; et al. From adenoma to CRC stages: The oral-gut microbiome axis as a source of potential microbial and metabolic biomarkers of malignancy. Neoplasia 2023, 40, 100901.
- Shi, Y.; Li, Z.; Zheng, W.; Liu, X.; Sun, C.; Laugsand, J.B.; Liu, Z.; Cui, G. Changes of immunocytic phenotypes and functions from human colorectal adenomatous stage to cancerous stage: Update. Immunobiology 2015, 220, 1186–1196.
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240.
- Wang, N.; Fang, J.Y. Fusobacterium nucleatum, a key pathogenic factor and microbial biomarker for colorectal cancer. Trends Microbiol. 2023, 31, 159–172.
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin. 2018, 68, 31–54.
- Song, M.; Chan, A.T. Environmental Factors, Gut Microbiota, and Colorectal Cancer Prevention. Clin. Gastroenterol. Hepatol. 2019, 17, 275–289.
- American Cancer Society, CRC Facts and Figures 2020–2022. 2020. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/colorectal-cancer-facts-and-figures/colorectal-cancer-facts-and-figures-2020-2022.pdf (accessed on 20 December 2022).
- Mariotto, A.B.; Yabroff, K.R.; Shao, Y.; Feuer, E.J.; Brown, M.L. Projections of the cost of cancer care in the United States: 2010–2020. J. Natl. Cancer Inst. 2011, 103, 117–128.
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254.
- Burnett-Hartman, A.N.; Lee, J.K.; Demb, J.; Gupta, S. An Update on the Epidemiology, Molecular Characterization, Diagnosis, and Screening Strategies for Early-Onset Colorectal Cancer. Gastroenterology 2021, 160, 1041–1049.
- American Institute for Cancer Research, World Cancer Research Fund, Diet, Nutrition, Physical Activity, and Colorectal Cancer. Continuous Update Projects (CUP). 2017. Available online: https://www.wcrf.org/wp-content/uploads/2021/02/Colorectal-cancer-report.pdf (accessed on 18 June 2018).
- Song, M.; Garrett, W.S.; Chan, A.T. Nutrients, foods, and colorectal cancer prevention. Gastroenterology 2015, 148, 1244–1260.e1216.
- Alrahawy, M.; Javed, S.; Atif, H.; Elsanhoury, K.; Mekhaeil, K.; Eskandar, G. Microbiome and Colorectal Cancer Management. Cureus 2022, 14, e30720.
- Bae, S.; Ulrich, C.M.; Neuhouser, M.L.; Malysheva, O.; Bailey, L.B.; Xiao, L.; Brown, E.C.; Cushing-Haugen, K.L.; Zheng, Y.; Cheng, T.Y.; et al. Plasma choline metabolites and colorectal cancer risk in the Women’s Health Initiative Observational Study. Cancer Res. 2014, 74, 7442–7452.
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorak, K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J. Gastroenterol. 2009, 15, 3329–3340.
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305.
- Yang, J.; Wei, H.; Zhou, Y.; Szeto, C.H.; Li, C.; Lin, Y.; Coker, O.O.; Lau, H.C.H.; Chan, A.W.H.; Sung, J.J.Y.; et al. High-Fat Diet Promotes Colorectal Tumorigenesis Through Modulating Gut Microbiota and Metabolites. Gastroenterology 2022, 162, 135–149.e132.
- Ocvirk, S.; O’Keefe, S.J.D. Dietary fat, bile acid metabolism and colorectal cancer. Semin. Cancer Biol. 2021, 73, 347–355.
- Murphy, N.; Norat, T.; Ferrari, P.; Jenab, M.; Bueno-de-Mesquita, B.; Skeie, G.; Dahm, C.C.; Overvad, K.; Olsen, A.; Tjønneland, A.; et al. Dietary fibre intake and risks of cancers of the colon and rectum in the European prospective investigation into cancer and nutrition (EPIC). PLoS ONE 2012, 7, e39361.
- Liu, P.H.; Wu, K.; Ng, K.; Zauber, A.G.; Nguyen, L.H.; Song, M.; He, X.; Fuchs, C.S.; Ogino, S.; Willett, W.C.; et al. Association of Obesity With Risk of Early-Onset Colorectal Cancer Among Women. JAMA Oncol. 2019, 5, 37–44.
- Greenblum, S.; Turnbaugh, P.J.; Borenstein, E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2012, 109, 594–599.
- Yang, B.; Bostick, R.M.; Tran, H.Q.; Gewirtz, A.T.; Campbell, P.T.; Fedirko, V. Circulating Biomarkers of Gut Barrier Function: Correlates and Nonresponse to Calcium Supplementation among Colon Adenoma Patients. Cancer Epidemiol. Biomark. Prev. 2016, 25, 318–326.
- Chen, R.; Luo, F.K.; Wang, Y.L.; Tang, J.L.; Liu, Y.S. LBP and CD14 polymorphisms correlate with increased colorectal carcinoma risk in Han Chinese. World J. Gastroenterol. 2011, 17, 2326–2331.
- Allen, J.M.; Mailing, L.J.; Niemiro, G.M.; Moore, R.; Cook, M.D.; White, B.A.; Holscher, H.D.; Woods, J.A. Exercise Alters Gut Microbiota Composition and Function in Lean and Obese Humans. Med. Sci. Sports Exerc. 2018, 50, 747–757.
- De Almeida, C.V.; de Camargo, M.R.; Russo, E.; Amedei, A. Role of diet and gut microbiota on colorectal cancer immunomodulation. World J. Gastroenterol. 2019, 25, 151–162.
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340.
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97.
- Huipeng, W.; Lifeng, G.; Chuang, G.; Jiaying, Z.; Yuankun, C. The differences in colonic mucosal microbiota between normal individual and colon cancer patients by polymerase chain reaction-denaturing gradient gel electrophoresis. J. Clin. Gastroenterol. 2014, 48, 138–144.
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum-symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166.
- Xu, W.; Zhou, W.; Wang, H.; Liang, S. Roles of Porphyromonas gingivalis and its virulence factors in periodontitis. Adv. Protein Chem. Struct. Biol. 2020, 120, 45–84.
- Rossi, O.; van Baarlen, P.; Wells, J.M. Host-recognition of pathogens and commensals in the mammalian intestine. Curr. Top. Microbiol. Immunol. 2013, 358, 291–321.
- Mantri, C.K.; Chen, C.H.; Dong, X.; Goodwin, J.S.; Pratap, S.; Paromov, V.; Xie, H. Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis. Microbiologyopen 2015, 4, 53–65.
- Xie, M.; Tang, Q.; Nie, J.; Zhang, C.; Zhou, X.; Yu, S.; Sun, J.; Cheng, X.; Dong, N.; Hu, Y.; et al. BMAL1-Downregulation Aggravates Porphyromonas Gingivalis-Induced Atherosclerosis by Encouraging Oxidative Stress. Circ. Res. 2020, 126, e15–e29.
- Wilensky, A.; Tzach-Nahman, R.; Potempa, J.; Shapira, L.; Nussbaum, G. Porphyromonas gingivalis gingipains selectively reduce CD14 expression, leading to macrophage hyporesponsiveness to bacterial infection. J. Innate Immun. 2015, 7, 127–135.
- Gaddis, D.E.; Maynard, C.L.; Weaver, C.T.; Michalek, S.M.; Katz, J. Role of TLR2-dependent IL-10 production in the inhibition of the initial IFN-γ T cell response to Porphyromonas gingivalis. J. Leukoc. Biol. 2013, 93, 21–31.
- Cerboni, S.; Gentili, M.; Manel, N. Diversity of pathogen sensors in dendritic cells. Adv. Immunol. 2013, 120, 211–237.
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22.
- Steinman, R.M. The control of immunity and tolerance by dendritic cell. Pathol. Biol. 2003, 51, 59–60.
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353.
- Luft, T.; Rodionova, E.; Maraskovsky, E.; Kirsch, M.; Hess, M.; Buchholtz, C.; Goerner, M.; Schnurr, M.; Skoda, R.; Ho, A.D. Adaptive functional differentiation of dendritic cells: Integrating the network of extra- and intracellular signals. Blood 2006, 107, 4763–4769.
- Kadowaki, N. Dendritic cells: A conductor of T cell differentiation. Allergol. Int. 2007, 56, 193–199.
- Jenkins, S.J.; Perona-Wright, G.; Worsley, A.G.; Ishii, N.; MacDonald, A.S. Dendritic cell expression of OX40 ligand acts as a costimulatory, not polarizing, signal for optimal Th2 priming and memory induction in vivo. J. Immunol. 2007, 179, 3515–3523.
- Huang, G.; Wang, Y.; Chi, H. Regulation of TH17 cell differentiation by innate immune signals. Cell Mol. Immunol. 2012, 9, 287–295.
- Levings, M.K.; Gregori, S.; Tresoldi, E.; Cazzaniga, S.; Bonini, C.; Roncarolo, M.G. Differentiation of Tr1 cells by immature dendritic cells requires IL-10 but not CD25+CD4+ Tr cells. Blood 2005, 105, 1162–1169.
- Sánchez-Paulete, A.R.; Teijeira, A.; Cueto, F.J.; Garasa, S.; Pérez-Gracia, J.L.; Sánchez-Arráez, A.; Sancho, D.; Melero, I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol. 2017, 28 (Suppl. S12), xii74.
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2018, 9, 3176.
- Rock, K.L.; Shen, L. Cross-presentation: Underlying mechanisms and role in immune surveillance. Immunol. Rev. 2005, 207, 166–183.
- Valladeau, J.; Ravel, O.; Dezutter-Dambuyant, C.; Moore, K.; Kleijmeer, M.; Liu, Y.; Duvert-Frances, V.; Vincent, C.; Schmitt, D.; Davoust, J.; et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 2000, 12, 71–81.
- Muzio, M.; Bosisio, D.; Polentarutti, N.; D’Amico, G.; Stoppacciaro, A.; Mancinelli, R.; van’t Veer, C.; Penton-Rol, G.; Ruco, L.P.; Allavena, P.; et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: Selective expression of TLR3 in dendritic cells. J. Immunol. 2000, 164, 5998–6004.
- Rescigno, M.; Granucci, F.; Ricciardi-Castagnoli, P. Molecular events of bacterial-induced maturation of dendritic cells. J. Clin. Immunol. 2000, 20, 161–166.
- Wilson, N.S.; El-Sukkari, D.; Villadangos, J.A. Dendritic cells constitutively present self antigens in their immature state in vivo and regulate antigen presentation by controlling the rates of MHC class II synthesis and endocytosis. Blood 2004, 103, 2187–2195.
- Deluce-Kakwata-Nkor, N.; Lamendour, L.; Chabot, V.; Héraud, A.; Ivanovic, Z.; Halary, F.; Dehaut, F.; Velge-Roussel, F. Differentiation of human dendritic cell subsets for immune tolerance induction. Transfus. Clin. Biol. 2018, 25, 90–95.
- Wallet, M.A.; Sen, P.; Flores, R.R.; Wang, Y.; Yi, Z.; Huang, Y.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Wang, B.; et al. MerTK is required for apoptotic cell-induced T cell tolerance. J. Exp. Med. 2008, 205, 219–232.
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215.
- Kaplan, C.W.; Ma, X.; Paranjpe, A.; Jewett, A.; Lux, R.; Kinder-Haake, S.; Shi, W. Fusobacterium nucleatum outer membrane proteins Fap2 and RadD induce cell death in human lymphocytes. Infect. Immun. 2010, 78, 4773–4778.
- Nosho, K.; Sukawa, Y.; Adachi, Y.; Ito, M.; Mitsuhashi, K.; Kurihara, H.; Kanno, S.; Yamamoto, I.; Ishigami, K.; Igarashi, H.; et al. Association of Fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J. Gastroenterol. 2016, 22, 557–566.
- Spigaglia, P.; Barbanti, F.; Germinario, E.A.P.; Criscuolo, E.M.; Bruno, G.; Sanchez-Mete, L.; Porowska, B.; Stigliano, V.; Accarpio, F.; Oddi, A.; et al. Comparison of microbiological profile of enterotoxigenic Bacteroides fragilis (ETBF) isolates from subjects with colorectal cancer (CRC) or intestinal pre-cancerous lesions versus healthy individuals and evaluation of environmental factors involved in intestinal dysbiosis. Anaerobe 2023, 82, 102757.
- Dejea, C.; Wick, E.; Sears, C.L. Bacterial oncogenesis in the colon. Future Microbiol. 2013, 8, 445–460.
- Ren, L.; Ye, J.; Zhao, B.; Sun, J.; Cao, P.; Yang, Y. The Role of Intestinal Microbiota in Colorectal Cancer. Front. Pharmacol. 2021, 12, 674807.
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785.
- American Cancer Society. Colorectal Cancer Early Detection, Diagnosis, and Staging. cancer.org | 1.800.227.2345. Available online: https://www.cancer.org/content/dam/CRC/PDF/Public/8606.00.pdf (accessed on 3 June 2022).
- Lochhead, P.; Chan, A.T.; Giovannucci, E.; Fuchs, C.S.; Wu, K.; Nishihara, R.; O’Brien, M.; Ogino, S. Progress and opportunities in molecular pathological epidemiology of colorectal premalignant lesions. Am. J. Gastroenterol. 2014, 109, 1205–1214.
- Frank, S.A. Incidence, Inheritance, and Evolution. In Dynamics of Cancer; Princeton University Press: Princeton, NJ, USA, 2007.
- Nagy, J.A.; Chang, S.H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869.
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072.
- Bateman, A.C. Pathology of serrated colorectal lesions. J. Clin. Pathol. 2014, 67, 865–874.
- Kang, M.; Edmundson, P.; Araujo-Perez, F.; McCoy, A.N.; Galanko, J.; Keku, T.O. Association of plasma endotoxin, inflammatory cytokines and risk of colorectal adenomas. BMC Cancer 2013, 13, 91.
- Melotte, V.; Lentjes, M.H.; van den Bosch, S.M.; Hellebrekers, D.M.; de Hoon, J.P.; Wouters, K.A.; Daenen, K.L.; Partouns-Hendriks, I.E.; Stessels, F.; Louwagie, J.; et al. N-Myc downstream-regulated gene 4 (NDRG4): A candidate tumor suppressor gene and potential biomarker for colorectal cancer. J. Natl. Cancer Inst. 2009, 101, 916–927.
- Yamane, L.; Scapulatempo-Neto, C.; Reis, R.M.; Guimarães, D.P. Serrated pathway in colorectal carcinogenesis. World J. Gastroenterol. 2014, 20, 2634–2640.
- Simon, K. Colorectal cancer development and advances in screening. Clin. Interv. Aging 2016, 11, 967–976.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Entry Collection:
Gastrointestinal Disease
Revisions:
2 times
(View History)
Update Date:
12 Dec 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No