Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jac A. Nickoloff | -- | 1871 | 2023-12-05 19:08:39 | | | |
| 2 | Jessie Wu | Meta information modification | 1871 | 2023-12-06 06:14:13 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nickoloff, J.A.; Jaiswal, A.S.; Sharma, N.; Williamson, E.A.; Tran, M.T.; Arris, D.; Yang, M.; Hromas, R. TATDN2. Encyclopedia. Available online: https://encyclopedia.pub/entry/52404 (accessed on 27 July 2026).
Nickoloff JA, Jaiswal AS, Sharma N, Williamson EA, Tran MT, Arris D, et al. TATDN2. Encyclopedia. Available at: https://encyclopedia.pub/entry/52404. Accessed July 27, 2026.
Nickoloff, Jac A., Aruna S. Jaiswal, Neelam Sharma, Elizabeth A. Williamson, Manh T. Tran, Dominic Arris, Ming Yang, Robert Hromas. "TATDN2" Encyclopedia, https://encyclopedia.pub/entry/52404 (accessed July 27, 2026).
Nickoloff, J.A., Jaiswal, A.S., Sharma, N., Williamson, E.A., Tran, M.T., Arris, D., Yang, M., & Hromas, R. (2023, December 05). TATDN2. In Encyclopedia. https://encyclopedia.pub/entry/52404
Nickoloff, Jac A., et al. "TATDN2." Encyclopedia. Web. 05 December, 2023.
Copy Citation
Replicative DNA polymerases are blocked by nearly all types of DNA damage. The resulting DNA replication stress threatens genome stability. DNA replication stress is also caused by depletion of nucleotide pools, DNA polymerase inhibitors, and DNA sequences or structures that are difficult to replicate. Replication stress triggers complex cellular responses that include cell cycle arrest, replication fork collapse to one-ended DNA double-strand breaks, induction of DNA repair, and programmed cell death after excessive damage. Replication stress caused by specific structures (e.g., G-rich sequences that form G-quadruplexes) is localized but occurs during the S phase of every cell division.
DNA damage
replication stress
oxidative DNA damage
genome instability
DNA double-strand breaks
structure-specific nucleases
1. Introduction
Cells respond to DNA damage by activating complex pathways that are collectively termed the DNA damage response (DDR). The DDR has emerged as a crucial network of damage sensing, signaling, and effector pathways that slow or stop cell cycle progression during each cell cycle phase and promote DNA repair and cell survival. However, when damage is excessive, hyperactivated DDR signaling can trigger programmed cell death by apoptosis or other cell death pathways [1][2][3][4][5]. These responses promote cell survival and genome stability in moderately damaged cells, and they eliminate excessively damaged cells, all of which contribute to tumor suppression. It is therefore not surprising that DDR factors are frequently inactivated in cancer, and DDR defects have been shown to destabilize the genome, leading to oncogene activation and/or tumor suppressor inactivation that drive tumorigenesis [4][6][7][8]. For example, in response to DNA damage, p53 plays critical roles in cell cycle checkpoint arrest, it stimulates many DNA repair pathways, and it regulates apoptosis [9][10][11]. Approximately half of tumors have p53 mutations, including inactivating mutations (revealing p53 as a tumor suppressor), and dominant negative (oncogenic gain-of-function) mutations that alter p53 tetramer activities [12]. More than 400 proteins are implicated in the DDR, but unlike p53, most are mutated in cancers at low frequencies [13][14]. The DDR is truly a double-edged sword: it maintains genome stability and suppresses tumorigenesis, but tumor cells can hijack the DDR to upregulate DNA repair and promote tumor cell survival in response to oncogenic stress and stress induced by genotoxic (DNA-damaging) chemotherapy and radiotherapy [15][16][17][18][19][20][21]. Tumors can also upregulate mutagenic repair pathways and this can promote rapid tumor evolution and therapeutic resistance [22][23][24][25]. The central role of the DDR in cancer etiology and treatment response accounts for the substantial ongoing efforts to describe mechanistic features of DDR factors and their attendant pathways, to identify specific DDR defects in tumors that have diagnostic and/or prognostic value, and to exploit DDR pathways and/or weaknesses due to tumor DDR defects to improve treatment of a broad range of cancers [4][6][26][27][28][29][30][31][32][33][34].
S-phase cells are more sensitive to DNA damage than cells in other cell cycle phases because most DNA lesions block DNA replication, and the consequent replication stress can cause lethal DNA double-strand breaks (DSBs) or genome rearrangements (i.e., dicentric chromosomes). DNA lesions that block replication include damaged bases such as those with open rings, chemical adducts (e.g., alkylated bases), oxidized bases, missing bases (apurinic/apyrimidinic sites), deaminated bases, and UV-induced pyrimidine dimers [35][36]. Most tumor cells divide rapidly and are more sensitive to genotoxins than normal tissues that divide infrequently or not at all, providing a therapeutic window for genotoxic chemotherapy and radiotherapy. Although genotoxic cancer therapeutics pose serious challenges due to associated side effects, these agents remain important treatment options, especially when targeted therapeutics are unavailable or tumors develop therapeutic resistance [37][38][39][40][41]. DNA damage interferes with DNA replication, causing replication forks to stall and triggering DNA replication stress, which activates replication stress response pathways that significantly overlap with DDR response pathways [42][43][44]. Thus, acute or chronic exposures to genotoxic chemicals or radiation cause widespread replication stress. Widespread replication stress can also be induced by DNA polymerase inhibitors, such as aphidicolin, and by reduced nucleotide pools [45][46]. Overactive cell growth pathways in cancer, caused for example by hyperactivated or overexpressed oncogenes, also induce widespread replication stress due to dysregulation of DNA replication timing and progression, a phenomenon termed oncogenic stress [47][48][49]. When RNA is generated during transcription, it can form stable RNA–DNA hybrids, termed R-loops, which can cause replication stress when encountered by replication machinery. Failure to properly process R-loops results in genome instability [50]. Researchers include R-loops in the widespread replication stress category because these sequences comprise roughly 5% of the human genome [51]. Even in the absence of DNA damage or other causes of widespread stress, replication stress is a normal feature of the S phase, particularly in metazoan cells with their large and complex genomes [52]. This is because certain DNA sequences are difficult to replicate, including minisatellite repeat sequences at common fragile sites; inverted repeats that can form double stem-loop (cruciform) structures; CAGn triplet repeats that form hairpin structures; purine-rich repeats (e.g., GAAn) that are capable of forming triple helical structures; G-rich DNA that can form stable looped, fold-back G-quadruplex structures; and telomere repeats that form branched structures at each end of linear chromosomes [53][54][55][56] (Figure 1). Such difficult-to-replicate sequences are often mutation hotspots, and they can be associated with translocations or other genome rearrangements in human disease, including neurological and developmental diseases, and cancer [57][58][59][60][61][62][63]. Researchers can thus characterize replication stress in two ways: widespread replication stress caused by DNA damage, inhibition of the replication machinery, or oncogenic stress; and localized replication stress at difficult-to-replicate sequences, which cells must manage during every S phase.
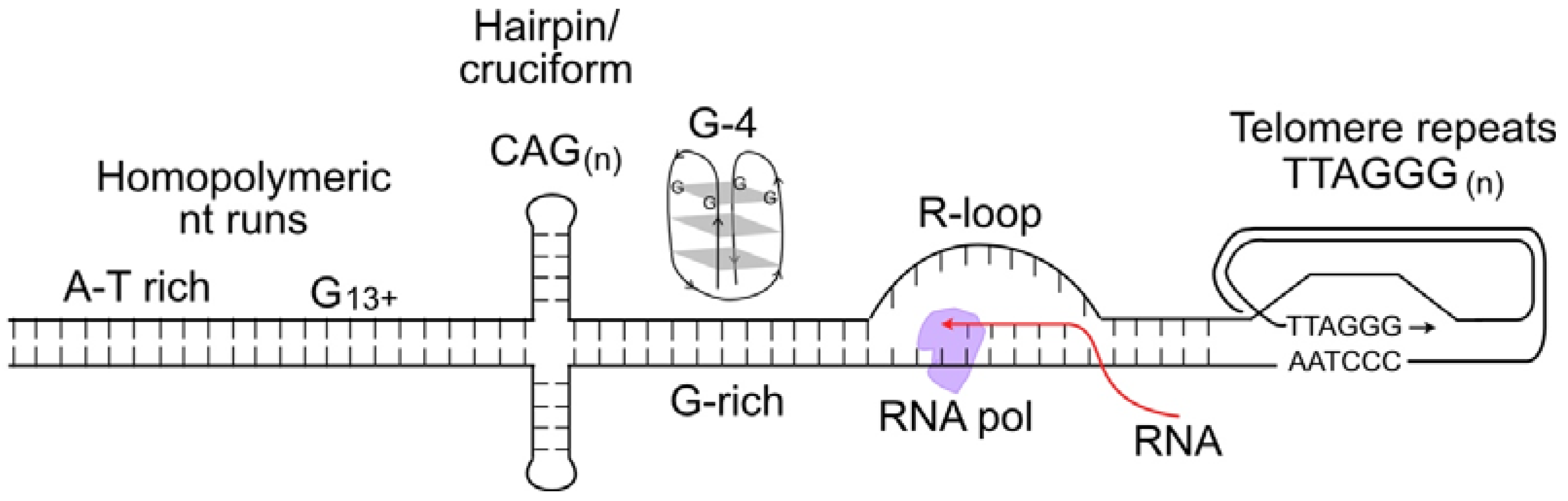
Figure 1. Nucleic acid structures that cause replication stress. These include difficult-to-replicate sequences such as homopolymeric nucleotide runs, palindromes and triplet repeats that can form stem-loop or cruciform structures, G-quadruplex DNA, and self-invading loops at telomeres. Stable R-loops cause replication stress when encountered by replicative DNA polymerases.
There are a variety of replication stress assays. Some assays monitor genome-wide replication such as nucleotide analog incorporation into DNA following pulse-labeling with readily detected thymidine analogs 5-ethynyl-2′-deoxyuridine (EdU), 5-chloro-2′-deoxyuridine (CldU), or bromo-deoxyuridine (BrdU); monitor phosphorylation/activation of checkpoint proteins Chk1, Chk2, ATR, and RPA; monitor phosphorylated histone H2AX (γ-H2AX), a marker of DSBs at collapsed forks; monitor cell cycle progression and cell cycle phase profiles; and monitor downstream effects of replication stress including comet assays that detect strand breaks at collapsed replication forks [64][65][66]. These endpoints can be assayed in cell populations using flow cytometry, or in individual cells using immunofluorescence microscopy to detect replication stress-induced nuclear foci. Checkpoint protein phosphorylation and γ-H2AX can also be detected using Western blot, or using proteomic mass spectrometry analysis [67]. Genome-wide replication stress can also be assayed with END-seq, a method that detects DSBs at nucleotide resolution [68][69]. Replication stress and replication fork restart are detected at the single-molecule level using DNA fiber analysis or DNA combing, which typically employ pulse-labeling with EdU, or dual pulse-labeling with EdU prior to stress induction, and CldU after release from stress [65][66][70][71][72]. Recruitment of DDR factors to stressed replication forks is detected using iPOND analysis, a type of chromatin immunoprecipitation assay [65][73][74]. These assays focus on cell responses in the S phase, but replication stress also generates single-stranded DNA (ssDNA) gaps detected beyond the S phase and even into subsequent cell cycles [75].
2. TATDN2 Is a Structure-Specific RNA Nuclease That Degrades RNA in R-Loops
R-loops typically form during RNA transcription and are a common and widespread source of replication stress. R-loops form when RNA stably pairs with the complementary DNA template strand, displacing the other DNA strand, creating a DNA bubble and RNA–DNA hybrid in a triple-stranded structure [76][77][78]. R-loops are evolutionarily conserved, and play important roles in chromatin remodeling, regulation of transcription, B-cell class switch recombination, RNA-mediated HR, and mitochondrial DNA replication [79][80][81][82][83][84][85][86]. R-loops frequently occur at promoter sequences, transcription termini, and enhancer and super-enhancer elements [77][78][79][80][87][88][89][90][91], and they also mediate recruitment of several factors that regulate histone modifications [78][90][91][92][93]. A critical normal function of R-loops is to relieve replication- or transcription-induced topological stress and, further, topoisomerase defects correlate with increased R-loop formation [94][95][96][97][98][99][100]. Despite these positive roles, R-loops also pose threats, including induction of DSBs, genome instability, and cancer, effects that are likely to reflect their propensity to induce replication stress [78][87][88][101][102][103][104][105][106][107]. One proposal is that replication fork encounters with stable R-loops cause fork collapse to a single-ended DSB leading to fork degradation, fork fusion, and chromosome translocations [105][106][107]. Other, not mutually exclusive, proposals are that the ssDNA in the R-loop DNA bubble is subject to nucleolytic attack [103], or that nucleases involved in transcription-coupled DNA repair cleave R-loop bubbles to induce DSBs [108].
Many proteins modulate R-loop formation and resolution. In yeast, the THO and THSC complexes prevent R-loop formation [109][110]. R-loops can be resolved by RNA unwinding by the helicases senataxin, aquarius (AQR), Werner syndrome protein (WRN), Bloom syndrome protein (BLM), regulator of telomere elongation helicase 1 (RTEL1), petite integration factor (PIF1), Fanconi anemia complementation group M (FANCM), alpha-thalassemia/mental retardation, X-linked (ATRX), and CRISPR-associated DinG protein (CasDinG) [111][112][113][114]. Several DEAD/H-box proteins including DDX1, DDX17, and DHX9 (RNA helicase A) are involved in both R-loop formation and resolution [115][116][117]. Proteins involved in homologous recombination and interstrand crosslink repair (FANCA, FANCD2) also function in R-loop resolution [118][119][120][121], and BRCA1 recruits senataxin to R-loops [83][122][123][124][125]. R-loops may also be resolved via RNA degradation by RNases, including RNaseH1 and RNaseH2 [126][127][128][129]. Deficiencies in R-loop resolution are pathological, evidenced by the several human diseases associated with defects in R-loop resolution factors, including amyotrophic lateral sclerosis type 4 and ataxia with oculomotor apraxia type 2 (senataxin) [111][112][113]; Fanconi anemia and various cancers (FANC proteins, BRCA1) [130]; adult-onset mitochondrial encephalomyopathy (RNase H1) [131]; and Aicardi–Goutières neurological syndrome (RNaseH2) [128].
Researchers recently investigated R-loop resolution in BRCA1-mutant cancer cells and found that these cells repress many microRNAs (miRs), probably due to overexpression of IRE1 RNase which degrades miRs involved in tumor suppression [132][133]. Re-expression of one of these miRs, miR-4638-5p, caused BRCA1-mutant cell death, but miR-4638-5p expression did not affect the viability of BRCA1 wild-type cells [66]. miR-4638-5p was found to repress TATDN2, one of three human paralogs of the conserved bacterial TatD nuclease family [134][135][136][137]. Researchers demonstrated that TATDN2 is a Mg2+-dependent, structure-specific 3′ RNA exonuclease and endonuclease that resolves R-loops via specific degradation of R-loop RNA [66] (Figure 2A). Downregulation of TATDN2 in BRCA1-mutant cells causes R-loop accumulation, decreased DNA replication, increased DSBs, genome instability, and cell death. Reconstituting BRCA1 in TATDN2-deficient cells suppressed all of these detrimental phenotypes. Together, these results indicate that the increased levels of R-loops in BRCA1-defective cells require TATDN2 for R-loop resolution to maintain genome integrity and cell viability. These results indicate that defects in both TATDN2 and BRCA1 are synthetically lethal (Figure 2B), which suggests novel therapeutic approaches to treat BRCA1-mutant tumors by targeting TATDN2, i.e., with small molecule inhibitors or via expression of miR-4638-5p [66].
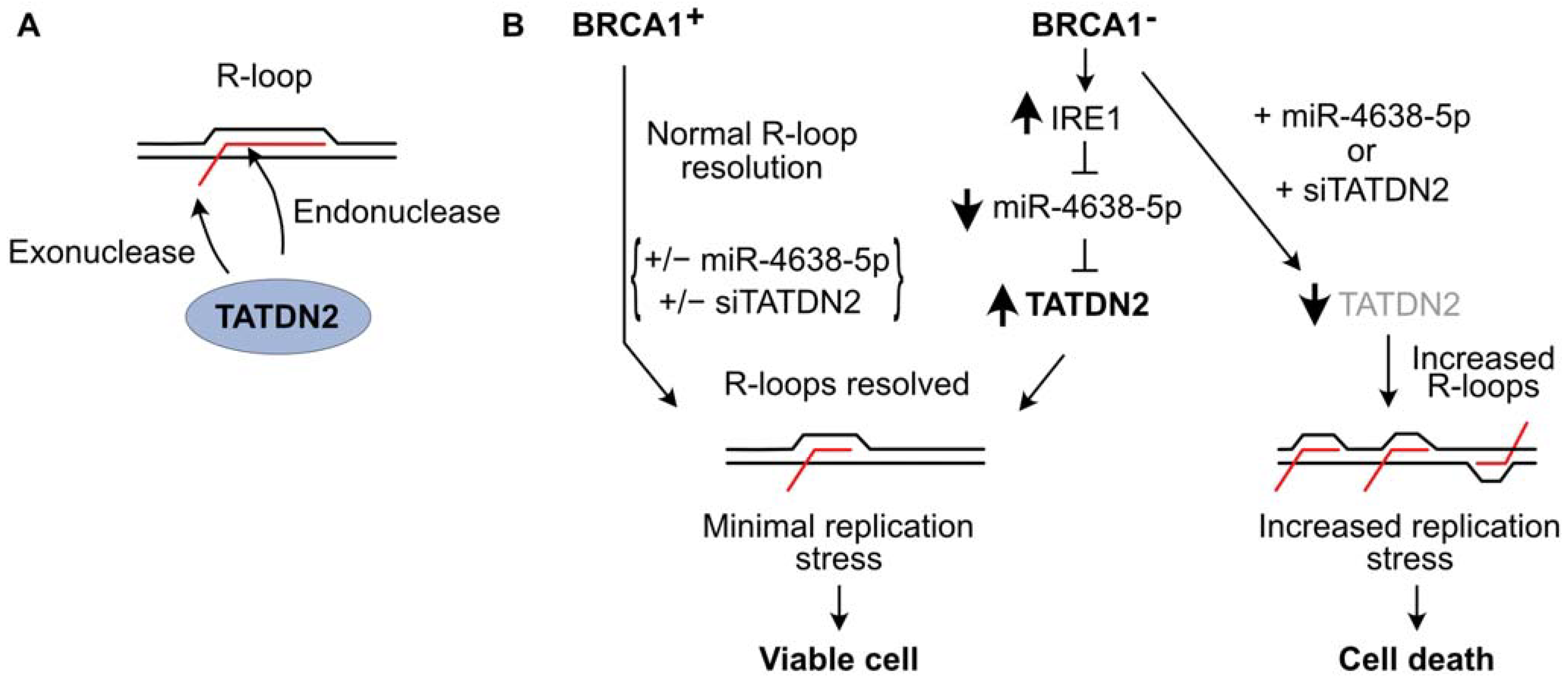
Figure 2. TATDN2 RNase promotes survival of BRCA1-defective cells by suppressing R-loop-induced replication stress. (A) TATDN2 is a structure-specific RNase that degrades RNA in R-loops with both exonuclease and endonuclease activities. (B) BRCA1 helps cells manage R-loops to prevent toxic replication stress. With functional BRCA1, adding or deleting miR-4638-5p or TATDN2 does not affect cell viability. In BRCA1-deficient cells, IRE1 RNase levels increase, reducing miR-4638-5p and increasing TATDN2 which acts to limit R-loops and associated replication stress, thereby promoting cell viability. Expressing miR-4638-5p or downregulating TATDN2 kills BRCA1-deficient cells due to increased R-loop-associated replication stress.
References
- Avalos, Y.; Canales, J.; Bravo-Sagua, R.; Criollo, A.; Lavandero, S.; Quest, A.F. Tumor suppression and promotion by autophagy. BioMed Res. Int. 2014, 2014, 603980.
- Carvajal, L.A.; Manfredi, J.J. Another fork in the road-life or death decisions by the tumour suppressor p53. EMBO Rep. 2013, 14, 414–421.
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248.
- Tian, H.; Gao, Z.; Li, H.; Zhang, B.; Wang, G.; Zhang, Q.; Pei, D.; Zheng, J. DNA damage response--a double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 2015, 358, 8–16.
- Molinaro, C.; Martoriati, A.; Cailliau, K. Proteins from the DNA damage response: Regulation, dysfunction, and anticancer strategies. Cancers 2021, 13, 3819.
- Datta, A.; Brosh, R.M., Jr. New insights into DNA helicases as druggable targets for cancer therapy. Front. Mol. Biosci. 2018, 5, 59.
- Jo, U.; Kim, H. Exploiting the Fanconi anemia pathway for targeted anti-cancer therapy. Mol. Cells 2015, 38, 669–676.
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870.
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070.
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113.
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104.
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8.
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560.
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180.
- Li, L.Y.; Guan, Y.D.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA repair pathways in cancer therapy and resistance. Front. Pharmacol. 2020, 11, 629266.
- Jurkovicova, D.; Neophytou, C.M.; Gasparovic, A.C.; Goncalves, A.C. DNA damage response in cancer therapy and resistance: Challenges and opportunities. Int. J. Mol. Sci. 2022, 23, 14672.
- Bertoli, C.; Herlihy, A.E.; Pennycook, B.R.; Kriston-Vizi, J.; de Bruin, R.A.M. Sustained E2F-dependent transcription Is a key mechanism to prevent replication-stress-induced DNA damage. Cell Rep. 2016, 15, 1412–1422.
- Belan, O.; Sebald, M.; Adamowicz, M.; Anand, R.; Vancevska, A.; Neves, J.; Grinkevich, V.; Hewitt, G.; Segura-Bayona, S.; Bellelli, R.; et al. POLQ seals post-replicative ssDNA gaps to maintain genome stability in BRCA-deficient cancer cells. Mol. Cell 2022, 82, 4664–4680.e4669.
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262.
- Wang, Z.; Jia, R.; Wang, L.; Yang, Q.; Hu, X.; Fu, Q.; Zhang, X.; Li, W.; Ren, Y. The emerging roles of Rad51 in cancer and its potential as a therapeutic target. Front. Oncol. 2022, 12, 935593.
- Beskow, C.; Skikuniene, J.; Holgersson, A.; Nilsson, B.; Lewensohn, R.; Kanter, L.; Viktorsson, K. Radioresistant cervical cancer shows upregulation of the NHEJ proteins DNA-PKcs, Ku70 and Ku86. Br. J. Cancer 2009, 101, 816–821.
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598.
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817.
- Willers, H.; Azzoli, C.G.; Santivasi, W.L.; Xia, F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013, 19, 200–207.
- Mikubo, M.; Inoue, Y.; Liu, G.; Tsao, M.S. Mechanism of drug tolerant persister cancer cells: The landscape and clinical implication for therapy. J. Thorac. Oncol. 2021, 16, 1798–1809.
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-deficient cancers provide new opportunities for precision oncology. Cancers 2020, 12, 687.
- Hromas, R.; Williamson, E.; Lee, S.H.; Nickoloff, J. Preventing the chromosomal translocations that cause cancer. Trans. Am. Clin. Climatol. Assoc. 2016, 127, 176–195.
- Nickoloff, J.A.; Jones, D.; Lee, S.-H.; Williamson, E.A.; Hromas, R. Drugging the cancers addicted to DNA repair. J. Natl. Cancer Inst. 2017, 109, djx059.
- Nickoloff, J.A. Toward greater precision in cancer radiotherapy. Cancer Res. 2021, 81, 3156–3157.
- Nickoloff, J.A.; Taylor, L.; Sharma, N.; Kato, T.A. Exploiting DNA repair pathways for tumor sensitization, mitigation of resistance, and normal tissue protection in radiotherapy. Cancer Drug Resist. 2021, 4, 244–263.
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176.
- Kamel, D.; Gray, C.; Walia, J.S.; Kumar, V. PARP inhibitor drugs in the treatment of breast, ovarian, prostate and pancreatic cancers: An update of clinical trials. Curr. Drug Targets 2018, 19, 21–37.
- Jannetti, S.A.; Zeglis, B.M.; Zalutsky, M.R.; Reiner, T. Poly(ADP-ribose)polymerase (PARP) inhibitors and radiation therapy. Front. Pharmacol. 2020, 11, 170.
- Luo, L.; Keyomarsi, K. PARP inhibitors as single agents and in combination therapy: The most promising treatment strategies in clinical trials for BRCA-mutant ovarian and triple-negative breast cancers. Expert Opin. Investig. Drugs 2022, 31, 607–631.
- Taylor, M.R.G.; Yeeles, J.T.P. The initial response of a eukaryotic replisome to DNA damage. Mol. Cell 2018, 70, 1067–1080.e1012.
- Friedberg, E.C.; Elledge, S.J.; Lehmann, A.R.; Lindahl, T.; Muzi-Falconi, M. (Eds.) DNA Repair, Mutagenesis, and Other Responses to DNA Damage, 1st ed.; Cold Spring Harbor Laboratory Press: Cold Sprng Harbor, NY, USA, 2014; p. 446.
- Morgan, M.A.; Lawrence, T.S. Molecular pathways: Overcoming radiation resistance by targeting DNA damage response pathways. Clin. Cancer Res. 2015, 21, 2898–2904.
- Stover, E.H.; Konstantinopoulos, P.A.; Matulonis, U.A.; Swisher, E.M. Biomarkers of response and resistance to DNA repair targeted therapies. Clin. Cancer Res. 2016, 22, 5651–5660.
- Masoud, V.; Pages, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017, 8, 120–134.
- Delou, J.M.A.; Souza, A.S.O.; Souza, L.C.M.; Borges, H.L. Highlights in resistance mechanism pathways for combination therapy. Cells 2019, 8, 1013.
- Pottier, C.; Fresnais, M.; Gilon, M.; Jerusalem, G.; Longuespee, R.; Sounni, N.E. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers 2020, 12, 731.
- Mognato, M.; Burdak-Rothkamm, S.; Rothkamm, K. Interplay between DNA replication stress, chromatin dynamics and DNA-damage response for the maintenance of genome stability. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108346.
- Liptay, M.; Barbosa, J.S.; Rottenberg, S. Replication fork remodeling and therapy escape in DNA damage response-deficient cancers. Front. Oncol. 2020, 10, 670.
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016, 37–38, 51–64.
- Mazouzi, A.; Stukalov, A.; Muller, A.C.; Chen, D.; Wiedner, M.; Prochazkova, J.; Chiang, S.C.; Schuster, M.; Breitwieser, F.P.; Pichlmair, A.; et al. A comprehensive analysis of the dynamic response to aphidicolin-mediated replication stress uncovers targets for ATM and ATMIN. Cell Rep. 2016, 15, 893–908.
- Singh, A.; Xu, Y.J. The cell killing mechanisms of hydroxyurea. Genes 2016, 7, 99.
- Hills, S.A.; Diffley, J.F. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014, 24, R435–R444.
- Primo, L.M.F.; Teixeira, L.K. DNA replication stress: Oncogenes in the spotlight. Genet. Mol. Biol. 2019, 43, e20190138.
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of oncogene-induced replication stress: Jigsaw falling into place. Cancer Discov. 2018, 8, 537–555.
- Stirling, P.C.; Chan, Y.A.; Minaker, S.W.; Aristizabal, M.J.; Barrett, I.; Sipahimalani, P.; Kobor, M.S.; Hieter, P. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev. 2012, 26, 163–175.
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chedin, F. Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals. Mol. Cell 2016, 63, 167–178.
- Rivera-Mulia, J.C.; Gilbert, D.M. Replicating large genomes: Divide and conquer. Mol. Cell 2016, 62, 756–765.
- Pearson, C.E.; Zorbas, H.; Price, G.B.; Zannis-Hadjopoulos, M. Inverted repeats, stem-loops, and cruciforms: Significance for initiation of DNA replication. J. Cell. Biochem. 1996, 63, 1–22.
- Jain, A.; Wang, G.; Vasquez, K.M. DNA triple helices: Biological consequences and therapeutic potential. Biochimie 2008, 90, 1117–1130.
- Miglietta, G.; Russo, M.; Capranico, G. G-quadruplex-R-loop interactions and the mechanism of anticancer G-quadruplex binders. Nucleic Acids Res. 2020, 48, 11942–11957.
- Aksenova, A.Y.; Mirkin, S.M. At the beginning of the end and in the middle of the beginning: Structure and maintenance of telomeric DNA repeats and interstitial telomeric sequences. Genes 2019, 10, 118.
- Ma, X.; Rogacheva, M.V.; Nishant, K.T.; Zanders, S.; Bustamante, C.D.; Alani, E. Mutation hot spots in yeast caused by long-range clustering of homopolymeric sequences. Cell Rep. 2012, 1, 36–42.
- Ma, K.; Qiu, L.; Mrasek, K.; Zhang, J.; Liehr, T.; Quintana, L.G.; Li, Z. Common fragile sites: Genomic hotspots of DNA damage and carcinogenesis. Int. J. Mol. Sci. 2012, 13, 11974–11999.
- Lemmens, B.; van Schendel, R.; Tijsterman, M. Mutagenic consequences of a single G-quadruplex demonstrate mitotic inheritance of DNA replication fork barriers. Nat. Commun. 2015, 6, 8909.
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501.
- Lu, R.; Pickett, H.A. Telomeric replication stress: The beginning and the end for alternative lengthening of telomeres cancers. Open Biol. 2022, 12, 220011.
- McDonald, M.J.; Yu, Y.H.; Guo, J.F.; Chong, S.Y.; Kao, C.F.; Leu, J.Y. Mutation at a distance caused by homopolymeric guanine repeats in Saccharomyces cerevisiae. Sci. Adv. 2016, 2, e1501033.
- Lee, W.T.C.; Yin, Y.; Morten, M.J.; Tonzi, P.; Gwo, P.P.; Odermatt, D.C.; Modesti, M.; Cantor, S.B.; Gari, K.; Huang, T.T.; et al. Single-molecule imaging reveals replication fork coupled formation of G-quadruplex structures hinders local replication stress signaling. Nat. Commun. 2021, 12, 2525.
- Saxena, S.; Zou, L. Hallmarks of DNA replication stress. Mol. Cell 2022, 82, 2298–2314.
- Wu, Y.; Lee, S.H.; Williamson, E.A.; Reinert, B.L.; Cho, J.H.; Xia, F.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Brantley, A.; et al. EEPD1 rescues stressed replication forks and maintains genome stability by promoting end resection and homologous recombination repair. PLoS Genet. 2015, 11, e1005675.
- Jaiswal, A.S.; Dutta, A.; Srinivasan, G.; Yuan, Y.; Zhou, D.; Shaheen, M.; Sadideen, D.T.; Kirby, A.; Williamson, E.A.; Gupta, Y.K.; et al. TATDN2 resolution of R-loops is required for survival of BRCA1-mutant cancer cells. Nucleic Acids Res. 2023, gkad952.
- Cortez, D. Proteomic analyses of the eukaryotic replication machinery. Methods Enzymol. 2017, 591, 33–53.
- Canela, A.; Sridharan, S.; Sciascia, N.; Tubbs, A.; Meltzer, P.; Sleckman, B.P.; Nussenzweig, A. DNA breaks and end resection measured genome-wide by end sequencing. Mol. Cell 2016, 63, 898–911.
- Tubbs, A.; Sridharan, S.; van Wietmarschen, N.; Maman, Y.; Callen, E.; Stanlie, A.; Wu, W.; Wu, X.; Day, A.; Wong, N.; et al. Dual roles of poly(dA:dT) tracts in replicationo initiation and fork collapse. Cell 2018, 174, 1127–1142.e119.
- Shimura, T.; Torres, M.J.; Martin, M.M.; Rao, V.A.; Pommier, Y.; Katsura, M.; Miyagawa, K.; Aladjem, M.I. Bloom’s syndrome helicase and Mus81 are required to induce transient double-strand DNA breaks in response to DNA replication stress. J. Mol. Biol. 2008, 375, 1152–1164.
- De Haro, L.P.; Wray, J.; Williamson, E.A.; Durant, S.T.; Corwin, L.; Gentry, A.C.; Osheroff, N.; Lee, S.H.; Hromas, R.; Nickoloff, J.A. Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res. 2010, 38, 5681–5691.
- Jaiswal, A.S.; Kim, H.S.; Scharer, O.D.; Sharma, N.; Williamson, E.A.; Srinivasan, G.; Phillips, L.; Kong, K.; Arya, S.; Misra, A.; et al. EEPD1 promotes repair of oxidatively-stressed replication forks. NAR Cancer 2023, 5, zcac044.
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327.
- Agudelo Garcia, P.A.; Gardner, M.; Freitas, M.A.; Parthun, M.R. Isolation of proteins on nascent chromatin and characterization by quantitative mass spectrometry. Methods Mol. Biol. 2019, 1983, 17–27.
- Quinet, A.; Carvajal-Maldonado, D.; Lemacon, D.; Vindigni, A. DNA fiber analysis: Mind the gap! Methods Enzymol. 2017, 591, 55–82.
- Thomas, M.; White, R.L.; Davis, R.W. Hybridization of RNA to double-stranded DNA: Formation of R-loops. Proc. Natl. Acad. Sci. USA 1976, 73, 2294–2298.
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178.
- Mackay, R.P.; Xu, Q.; Weinberger, P.M. R-loop physiology and pathology: A brief review. DNA Cell Biol. 2020, 39, 1914–1925.
- Chen, P.B.; Chen, H.V.; Acharya, D.; Rando, O.J.; Fazzio, T.G. R loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat. Struct. Mol. Biol. 2015, 22, 999–1007.
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825.
- Tian, M.; Alt, F.W. Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. J. Biol. Chem. 2000, 275, 24163–24172.
- So, C.C.; Martin, A. DSB structure impacts DNA recombination leading to class switching and chromosomal translocations in human B cells. PLoS Genet. 2019, 15, e1008101.
- Teng, Y.; Yadav, T.; Duan, M.; Tan, J.; Xiang, Y.; Gao, B.; Xu, J.; Liang, Z.; Liu, Y.; Nakajima, S.; et al. ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat. Commun. 2018, 9, 4115.
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell 2018, 175, 558–570.e511.
- Holt, I.J. R-loops and mitochondrial DNA metabolism. Methods Mol. Biol. 2022, 2528, 173–202.
- Posse, V.; Al-Behadili, A.; Uhler, J.P.; Clausen, A.R.; Reyes, A.; Zeviani, M.; Falkenberg, M.; Gustafsson, C.M. RNase H1 directs origin-specific initiation of DNA replication in human mitochondria. PLoS Genet. 2019, 15, e1007781.
- Rinaldi, C.; Pizzul, P.; Longhese, M.P.; Bonetti, D. Sensing R-loop-associated DNA damage to safeguard genome stability. Front. Cell Dev. Biol. 2020, 8, 618157.
- Brickner, J.R.; Garzon, J.L.; Cimprich, K.A. Walking a tightrope: The complex balancing act of R-loops in genome stability. Mol. Cell 2022, 82, 2267–2297.
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chedin, F. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600.
- Castellano-Pozo, M.; Santos-Pereira, J.M.; Rondon, A.G.; Barroso, S.; Andujar, E.; Perez-Alegre, M.; Garcia-Muse, T.; Aguilera, A. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 2013, 52, 583–590.
- Zhou, H.; Li, L.; Wang, Q.; Hu, Y.; Zhao, W.; Gautam, M.; Li, L. H3K9 demethylation-induced R-loop accumulation is linked to disorganized nucleoli. Front. Genet. 2020, 11, 43.
- Fazzio, T.G. Regulation of chromatin structure and cell fate by R-loops. Transcription 2016, 7, 121–126.
- Chedin, F. Nascent connections: R-loops and chromatin patterning. Trends Genet. 2016, 32, 828–838.
- Drolet, M.; Bi, X.; Liu, L.F. Hypernegative supercoiling of the DNA template during transcription elongation in vitro. J. Biol. Chem. 1994, 269, 2068–2074.
- Drolet, M. Growth inhibition mediated by excess negative supercoiling: The interplay between transcription elongation, R-loop formation and DNA topology. Mol. Microbiol. 2006, 59, 723–730.
- Stolz, R.; Sulthana, S.; Hartono, S.R.; Malig, M.; Benham, C.J.; Chedin, F. Interplay between DNA sequence and negative superhelicity drives R-loop structures. Proc. Natl. Acad. Sci. USA 2019, 116, 6260–6269.
- Phoenix, P.; Raymond, M.A.; Masse, E.; Drolet, M. Roles of DNA topoisomerases in the regulation of R-loop formation in vitro. J. Biol. Chem. 1997, 272, 1473–1479.
- El Hage, A.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558.
- Manzo, S.G.; Hartono, S.R.; Sanz, L.A.; Marinello, J.; De Biasi, S.; Cossarizza, A.; Capranico, G.; Chedin, F. DNA Topoisomerase I differentially modulates R-loops across the human genome. Genome Biol. 2018, 19, 100.
- Masse, E.; Drolet, M. Escherichia coli DNA topoisomerase I inhibits R-loop formation by relaxing transcription-induced negative supercoiling. J. Biol. Chem. 1999, 274, 16659–16664.
- Wahba, L.; Gore, S.K.; Koshland, D. The homologous recombination machinery modulates the formation of RNA-DNA hybrids and associated chromosome instability. Elife 2013, 2, e00505.
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396.
- Wimberly, H.; Shee, C.; Thornton, P.C.; Sivaramakrishnan, P.; Rosenberg, S.M.; Hastings, P.J. R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli. Nat. Commun. 2013, 4, 2115.
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056.
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977.
- Lang, K.S.; Hall, A.N.; Merrikh, C.N.; Ragheb, M.; Tabakh, H.; Pollock, A.J.; Woodward, J.J.; Dreifus, J.E.; Merrikh, H. Replication-transcription conflicts generate R-loops that orchestrate bacterial stress survival and pathogenesis. Cell 2017, 170, 787–799.e718.
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 2017, 170, 774–786.e719.
- Roy, D.; Zhang, Z.; Lu, Z.; Hsieh, C.L.; Lieber, M.R. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: A nick can serve as a strong R-loop initiation site. Mol. Cell. Biol. 2010, 30, 146–159.
- San Martin-Alonso, M.; Soler-Oliva, M.E.; Garcia-Rubio, M.; Garcia-Muse, T.; Aguilera, A. Harmful R-loops are prevented via different cell cycle-specific mechanisms. Nat. Commun. 2021, 12, 4451.
- Gonzalez-Aguilera, C.; Tous, C.; Gomez-Gonzalez, B.; Huertas, P.; Luna, R.; Aguilera, A. The THP1-SAC3-SUS1-CDC31 complex works in transcription elongation-mRNA export preventing RNA-mediated genome instability. Mol. Biol. Cell 2008, 19, 4310–4318.
- Moreira, M.C.; Klur, S.; Watanabe, M.; Nemeth, A.H.; Le Ber, I.; Moniz, J.C.; Tranchant, C.; Aubourg, P.; Tazir, M.; Schols, L.; et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004, 36, 225–227.
- Cohen, S.; Puget, N.; Lin, Y.L.; Clouaire, T.; Aguirrebengoa, M.; Rocher, V.; Pasero, P.; Canitrot, Y.; Legube, G. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018, 9, 533.
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805.
- Tran, P.L.T.; Pohl, T.J.; Chen, C.F.; Chan, A.; Pott, S.; Zakian, V.A. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nat. Commun. 2017, 8, 15025.
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Rep. 2018, 23, 1891–1905.
- Yuan, W.; Al-Hadid, Q.; Wang, Z.; Shen, L.; Cho, H.; Wu, X.; Yang, Y. TDRD3 promotes DHX9 chromatin recruitment and R-loop resolution. Nucleic Acids Res. 2021, 49, 8573–8591.
- Yang, S.; Winstone, L.; Mondal, S.; Wu, Y. Helicases in R-loop formation and resolution. J. Biol. Chem. 2023, 299, 105307.
- Pan, X.; Chen, Y.; Biju, B.; Ahmed, N.; Kong, J.; Goldenberg, M.; Huang, J.; Mohan, N.; Klosek, S.; Parsa, K.; et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep. 2019, 9, 19110.
- Garcia-Rubio, M.L.; Perez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi anemia pathway protects genome integrity from R-loops. PLoS Genet. 2015, 11, e1005674.
- Okamoto, Y.; Abe, M.; Itaya, A.; Tomida, J.; Ishiai, M.; Takaori-Kondo, A.; Taoka, M.; Isobe, T.; Takata, M. FANCD2 protects genome stability by recruiting RNA processing enzymes to resolve R-loops during mild replication stress. FEBS J. 2019, 286, 139–150.
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell 2015, 60, 351–361.
- San Martin Alonso, M.; Noordermeer, S.M. Untangling the crosstalk between BRCA1 and R-loops during DNA repair. Nucleic Acids Res. 2021, 49, 4848–4863.
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365.
- Wang, Y.; Ma, B.; Liu, X.; Gao, G.; Che, Z.; Fan, M.; Meng, S.; Zhao, X.; Sugimura, R.; Cao, H.; et al. ZFP281-BRCA2 prevents R-loop accumulation during DNA replication. Nat. Commun. 2022, 13, 3493.
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647.
- Lima, W.F.; Murray, H.M.; Damle, S.S.; Hart, C.E.; Hung, G.; De Hoyos, C.L.; Liang, X.H.; Crooke, S.T. Viable RNaseH1 knockout mice show RNaseH1 is essential for R loop processing, mitochondrial and liver function. Nucleic Acids Res. 2016, 44, 5299–5312.
- Cornelio, D.A.; Sedam, H.N.; Ferrarezi, J.A.; Sampaio, N.M.; Argueso, J.L. Both R-loop removal and ribonucleotide excision repair activities of RNase H2 contribute substantially to chromosome stability. DNA Repair 2017, 52, 110–114.
- Cristini, A.; Tellier, M.; Constantinescu, F.; Accalai, C.; Albulescu, L.O.; Heiringhoff, R.; Bery, N.; Sordet, O.; Murphy, S.; Gromak, N. RNase H2, mutated in Aicardi-Goutieres syndrome, resolves co-transcriptional R-loops to prevent DNA breaks and inflammation. Nat. Commun. 2022, 13, 2961.
- Nguyen, H.D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T.A.; Zou, L. Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol. Cell 2017, 65, 832–847.e834.
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349.
- Reyes, A.; Melchionda, L.; Nasca, A.; Carrara, F.; Lamantea, E.; Zanolini, A.; Lamperti, C.; Fang, M.; Zhang, J.; Ronchi, D.; et al. RNASEH1 mutations Impair mtDNA replication and cause adult-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 2015, 97, 186–193.
- Hromas, R.; Srinivasan, G.; Yang, M.; Jaiswal, A.; Totterdale, T.A.; Phillips, L.; Kirby, A.; Khodayari, N.; Brantley, M.; Williamson, E.A.; et al. BRCA1 mediates protein homeostasis through the ubiquitination of PERK and IRE1. iScience 2022, 25, 105626.
- Zhang, K.; Liu, H.; Song, Z.; Jiang, Y.; Kim, H.; Samavati, L.; Nguyen, H.M.; Yang, Z.Q. The UPR transducer IRE1 promotes breast cancer malignancy by degrading tumor suppressor microRNAs. iScience 2020, 23, 101503.
- Lee, K.Y.; Cheon, S.H.; Kim, D.G.; Lee, S.J.; Lee, B.J. A structural study of TatD from Staphylococcus aureus elucidates a putative DNA-binding mode of a Mg2+-dependent nuclease. IUCrJ 2020, 7, 509–521.
- Chen, Y.C.; Li, C.L.; Hsiao, Y.Y.; Duh, Y.; Yuan, H.S. Structure and function of TatD exonuclease in DNA repair. Nucleic Acids Res. 2014, 42, 10776–10785.
- Singh, D.; Rahi, A.; Kumari, R.; Gupta, V.; Gautam, G.; Aggarwal, S.; Rehan, M.; Bhatnagar, R. Computational and mutational analysis of TatD DNase of Bacillus anthracis. J. Cell. Biochem. 2019, 120, 11318–11330.
- Dorival, J.; Eichman, B.F. Human and bacterial TatD enzymes exhibit apurinic/apyrimidinic (AP) endonuclease activity. Nucleic Acids Res. 2023, 51, 2838–2849.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
855
Revisions:
2 times
(View History)
Update Date:
06 Dec 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No