Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jac A. Nickoloff and Version 2 by Jessie Wu.
Replicative DNA polymerases are blocked by nearly all types of DNA damage. The resulting DNA replication stress threatens genome stability. DNA replication stress is also caused by depletion of nucleotide pools, DNA polymerase inhibitors, and DNA sequences or structures that are difficult to replicate. Replication stress triggers complex cellular responses that include cell cycle arrest, replication fork collapse to one-ended DNA double-strand breaks, induction of DNA repair, and programmed cell death after excessive damage. Replication stress caused by specific structures (e.g., G-rich sequences that form G-quadruplexes) is localized but occurs during the S phase of every cell division.
- DNA damage
- replication stress
- oxidative DNA damage
- genome instability
- DNA double-strand breaks
- structure-specific nucleases
1. Introduction
Cells respond to DNA damage by activating complex pathways that are collectively termed the DNA damage response (DDR). The DDR has emerged as a crucial network of damage sensing, signaling, and effector pathways that slow or stop cell cycle progression during each cell cycle phase and promote DNA repair and cell survival. However, when damage is excessive, hyperactivated DDR signaling can trigger programmed cell death by apoptosis or other cell death pathways [1][2][3][4][5][1,2,3,4,5]. These responses promote cell survival and genome stability in moderately damaged cells, and they eliminate excessively damaged cells, all of which contribute to tumor suppression. It is therefore not surprising that DDR factors are frequently inactivated in cancer, and DDR defects have been shown to destabilize the genome, leading to oncogene activation and/or tumor suppressor inactivation that drive tumorigenesis [4][6][7][8][4,6,7,8]. For example, in response to DNA damage, p53 plays critical roles in cell cycle checkpoint arrest, it stimulates many DNA repair pathways, and it regulates apoptosis [9][10][11][9,10,11]. Approximately half of tumors have p53 mutations, including inactivating mutations (revealing p53 as a tumor suppressor), and dominant negative (oncogenic gain-of-function) mutations that alter p53 tetramer activities [12]. More than 400 proteins are implicated in the DDR, but unlike p53, most are mutated in cancers at low frequencies [13][14][13,14]. The DDR is truly a double-edged sword: it maintains genome stability and suppresses tumorigenesis, but tumor cells can hijack the DDR to upregulate DNA repair and promote tumor cell survival in response to oncogenic stress and stress induced by genotoxic (DNA-damaging) chemotherapy and radiotherapy [15][16][17][18][19][20][21][15,16,17,18,19,20,21]. Tumors can also upregulate mutagenic repair pathways and this can promote rapid tumor evolution and therapeutic resistance [22][23][24][25][22,23,24,25]. The central role of the DDR in cancer etiology and treatment response accounts for the substantial ongoing efforts to describe mechanistic features of DDR factors and their attendant pathways, to identify specific DDR defects in tumors that have diagnostic and/or prognostic value, and to exploit DDR pathways and/or weaknesses due to tumor DDR defects to improve treatment of a broad range of cancers [4][6][26][27][28][29][30][31][32][33][34][4,6,26,27,28,29,30,31,32,33,34].
S-phase cells are more sensitive to DNA damage than cells in other cell cycle phases because most DNA lesions block DNA replication, and the consequent replication stress can cause lethal DNA double-strand breaks (DSBs) or genome rearrangements (i.e., dicentric chromosomes). DNA lesions that block replication include damaged bases such as those with open rings, chemical adducts (e.g., alkylated bases), oxidized bases, missing bases (apurinic/apyrimidinic sites), deaminated bases, and UV-induced pyrimidine dimers [35][36][35,36]. Most tumor cells divide rapidly and are more sensitive to genotoxins than normal tissues that divide infrequently or not at all, providing a therapeutic window for genotoxic chemotherapy and radiotherapy. Although genotoxic cancer therapeutics pose serious challenges due to associated side effects, these agents remain important treatment options, especially when targeted therapeutics are unavailable or tumors develop therapeutic resistance [37][38][39][40][41][37,38,39,40,41]. DNA damage interferes with DNA replication, causing replication forks to stall and triggering DNA replication stress, which activates replication stress response pathways that significantly overlap with DDR response pathways [42][43][44][42,43,44]. Thus, acute or chronic exposures to genotoxic chemicals or radiation cause widespread replication stress. Widespread replication stress can also be induced by DNA polymerase inhibitors, such as aphidicolin, and by reduced nucleotide pools [45][46][45,46]. Overactive cell growth pathways in cancer, caused for example by hyperactivated or overexpressed oncogenes, also induce widespread replication stress due to dysregulation of DNA replication timing and progression, a phenomenon termed oncogenic stress [47][48][49][47,48,49]. When RNA is generated during transcription, it can form stable RNA–DNA hybrids, termed R-loops, which can cause replication stress when encountered by replication machinery. Failure to properly process R-loops results in genome instability [50]. RWesearchers include R-loops in the widespread replication stress category because these sequences comprise roughly 5% of the human genome [51]. Even in the absence of DNA damage or other causes of widespread stress, replication stress is a normal feature of the S phase, particularly in metazoan cells with their large and complex genomes [52]. This is because certain DNA sequences are difficult to replicate, including minisatellite repeat sequences at common fragile sites; inverted repeats that can form double stem-loop (cruciform) structures; CAGn triplet repeats that form hairpin structures; purine-rich repeats (e.g., GAAn) that are capable of forming triple helical structures; G-rich DNA that can form stable looped, fold-back G-quadruplex structures; and telomere repeats that form branched structures at each end of linear chromosomes [53][54][55][56][53,54,55,56] (Figure 1). Such difficult-to-replicate sequences are often mutation hotspots, and they can be associated with translocations or other genome rearrangements in human disease, including neurological and developmental diseases, and cancer [57][58][59][60][61][62][63][57,58,59,60,61,62,63]. RWesearchers can thus characterize replication stress in two ways: widespread replication stress caused by DNA damage, inhibition of the replication machinery, or oncogenic stress; and localized replication stress at difficult-to-replicate sequences, which cells must manage during every S phase.
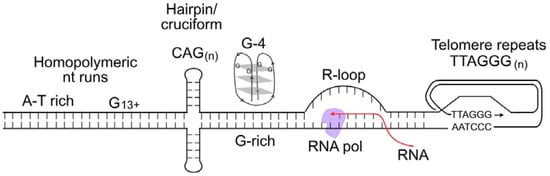
Figure 1. Nucleic acid structures that cause replication stress. These include difficult-to-replicate sequences such as homopolymeric nucleotide runs, palindromes and triplet repeats that can form stem-loop or cruciform structures, G-quadruplex DNA, and self-invading loops at telomeres. Stable R-loops cause replication stress when encountered by replicative DNA polymerases.
There are a variety of replication stress assays. Some assays monitor genome-wide replication such as nucleotide analog incorporation into DNA following pulse-labeling with readily detected thymidine analogs 5-ethynyl-2′-deoxyuridine (EdU), 5-chloro-2′-deoxyuridine (CldU), or bromo-deoxyuridine (BrdU); monitor phosphorylation/activation of checkpoint proteins Chk1, Chk2, ATR, and RPA; monitor phosphorylated histone H2AX (γ-H2AX), a marker of DSBs at collapsed forks; monitor cell cycle progression and cell cycle phase profiles; and monitor downstream effects of replication stress including comet assays that detect strand breaks at collapsed replication forks [64][65][66][64,65,66]. These endpoints can be assayed in cell populations using flow cytometry, or in individual cells using immunofluorescence microscopy to detect replication stress-induced nuclear foci. Checkpoint protein phosphorylation and γ-H2AX can also be detected using Western blot, or using proteomic mass spectrometry analysis [67]. Genome-wide replication stress can also be assayed with END-seq, a method that detects DSBs at nucleotide resolution [68][69][68,69]. Replication stress and replication fork restart are detected at the single-molecule level using DNA fiber analysis or DNA combing, which typically employ pulse-labeling with EdU, or dual pulse-labeling with EdU prior to stress induction, and CldU after release from stress [65][66][70][71][72][65,66,70,71,72]. Recruitment of DDR factors to stressed replication forks is detected using iPOND analysis, a type of chromatin immunoprecipitation assay [65][73][74][65,73,74]. These assays focus on cell responses in the S phase, but replication stress also generates single-stranded DNA (ssDNA) gaps detected beyond the S phase and even into subsequent cell cycles [75].
2. TATDN2 Is a Structure-Specific RNA Nuclease That Degrades RNA in R-Loops
R-loops typically form during RNA transcription and are a common and widespread source of replication stress. R-loops form when RNA stably pairs with the complementary DNA template strand, displacing the other DNA strand, creating a DNA bubble and RNA–DNA hybrid in a triple-stranded structure [76][77][78][212,213,214]. R-loops are evolutionarily conserved, and play important roles in chromatin remodeling, regulation of transcription, B-cell class switch recombination, RNA-mediated HR, and mitochondrial DNA replication [79][80][81][82][83][84][85][86][215,216,217,218,219,220,221,222]. R-loops frequently occur at promoter sequences, transcription termini, and enhancer and super-enhancer elements [77][78][79][80][87][88][89][90][91][213,214,215,216,223,224,225,226,227], and they also mediate recruitment of several factors that regulate histone modifications [78][90][91][92][93][214,226,227,228,229]. A critical normal function of R-loops is to relieve replication- or transcription-induced topological stress and, further, topoisomerase defects correlate with increased R-loop formation [94][95][96][97][98][99][100][230,231,232,233,234,235,236]. Despite these positive roles, R-loops also pose threats, including induction of DSBs, genome instability, and cancer, effects that are likely to reflect their propensity to induce replication stress [78][87][88][101][102][103][104][105][106][107][214,223,224,237,238,239,240,241,242,243]. One proposal is that replication fork encounters with stable R-loops cause fork collapse to a single-ended DSB leading to fork degradation, fork fusion, and chromosome translocations [105][106][107][241,242,243]. Other, not mutually exclusive, proposals are that the ssDNA in the R-loop DNA bubble is subject to nucleolytic attack [103][239], or that nucleases involved in transcription-coupled DNA repair cleave R-loop bubbles to induce DSBs [108][244].
Many proteins modulate R-loop formation and resolution. In yeast, the THO and THSC complexes prevent R-loop formation [109][110][245,246]. R-loops can be resolved by RNA unwinding by the helicases senataxin, aquarius (AQR), Werner syndrome protein (WRN), Bloom syndrome protein (BLM), regulator of telomere elongation helicase 1 (RTEL1), petite integration factor (PIF1), Fanconi anemia complementation group M (FANCM), alpha-thalassemia/mental retardation, X-linked (ATRX), and CRISPR-associated DinG protein (CasDinG) [111][112][113][114][247,248,249,250]. Several DEAD/H-box proteins including DDX1, DDX17, and DHX9 (RNA helicase A) are involved in both R-loop formation and resolution [115][116][117][251,252,253]. Proteins involved in homologous recombination and interstrand crosslink repair (FANCA, FANCD2) also function in R-loop resolution [118][119][120][121][254,255,256,257], and BRCA1 recruits senataxin to R-loops [83][122][123][124][125][219,258,259,260,261]. R-loops may also be resolved via RNA degradation by RNases, including RNaseH1 and RNaseH2 [126][127][128][129][262,263,264,265]. Deficiencies in R-loop resolution are pathological, evidenced by the several human diseases associated with defects in R-loop resolution factors, including amyotrophic lateral sclerosis type 4 and ataxia with oculomotor apraxia type 2 (senataxin) [111][112][113][247,248,249]; Fanconi anemia and various cancers (FANC proteins, BRCA1) [130][266]; adult-onset mitochondrial encephalomyopathy (RNase H1) [131][267]; and Aicardi–Goutières neurological syndrome (RNaseH2) [128][264].
RWesearchers recently investigated R-loop resolution in BRCA1-mutant cancer cells and found that these cells repress many microRNAs (miRs), probably due to overexpression of IRE1 RNase which degrades miRs involved in tumor suppression [132][133][268,269]. Re-expression of one of these miRs, miR-4638-5p, caused BRCA1-mutant cell death, but miR-4638-5p expression did not affect the viability of BRCA1 wild-type cells [66]. miR-4638-5p was found to repress TATDN2, one of three human paralogs of the conserved bacterial TatD nuclease family [134][135][136][137][270,271,272,273]. RWesearchers demonstrated that TATDN2 is a Mg2+-dependent, structure-specific 3′ RNA exonuclease and endonuclease that resolves R-loops via specific degradation of R-loop RNA [66] (Figure 2A). Downregulation of TATDN2 in BRCA1-mutant cells causes R-loop accumulation, decreased DNA replication, increased DSBs, genome instability, and cell death. Reconstituting BRCA1 in TATDN2-deficient cells suppressed all of these detrimental phenotypes. Together, these results indicate that the increased levels of R-loops in BRCA1-defective cells require TATDN2 for R-loop resolution to maintain genome integrity and cell viability. These results indicate that defects in both TATDN2 and BRCA1 are synthetically lethal (Figure 2B), which suggests novel therapeutic approaches to treat BRCA1-mutant tumors by targeting TATDN2, i.e., with small molecule inhibitors or via expression of miR-4638-5p [66].
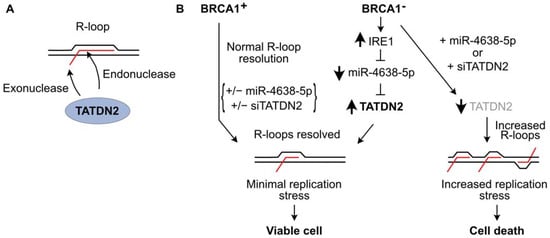
Figure 2. TATDN2 RNase promotes survival of BRCA1-defective cells by suppressing R-loop-induced replication stress. (A) TATDN2 is a structure-specific RNase that degrades RNA in R-loops with both exonuclease and endonuclease activities. (B) BRCA1 helps cells manage R-loops to prevent toxic replication stress. With functional BRCA1, adding or deleting miR-4638-5p or TATDN2 does not affect cell viability. In BRCA1-deficient cells, IRE1 RNase levels increase, reducing miR-4638-5p and increasing TATDN2 which acts to limit R-loops and associated replication stress, thereby promoting cell viability. Expressing miR-4638-5p or downregulating TATDN2 kills BRCA1-deficient cells due to increased R-loop-associated replication stress.