 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Minzhong Yu | -- | 2860 | 2023-11-23 14:25:24 | | | |
| 2 | Mona Zou | -2 word(s) | 2858 | 2023-11-27 09:18:32 | | |
Video Upload Options
Inherited retinal dystrophies encompass a diverse group of disorders affecting the structure and function of the retina, leading to progressive visual impairment and, in severe cases, blindness. Electrophysiology testing has emerged as a valuable tool in assessing and diagnosing those conditions, offering insights into the function of different parts of the visual pathway from retina to visual cortex and aiding in disease classification. The different applications and limitations of electrophysiology techniques, including multifocal electroretinogram (mfERG), full-field ERG (ffERG), electrooculogram (EOG), pattern electroretinogram (PERG), and visual evoked potential (VEP), in the diagnosis and management of these distinctive phenotypes are discussed.
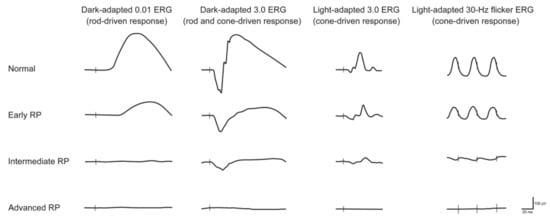
1. Retinitis Pigmentosa
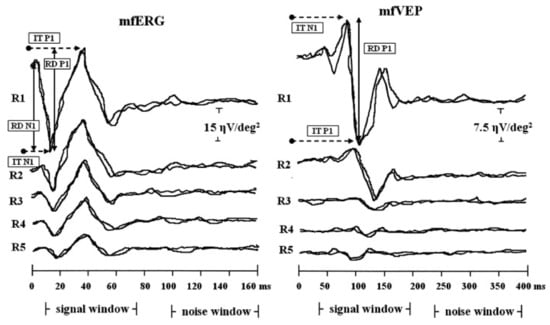
2. Progressive Cone/Cone-Rod Dystrophy
3. Cone Dystrophy with Supernormal Rod Response
4. Enhanced S-Cone Syndrome
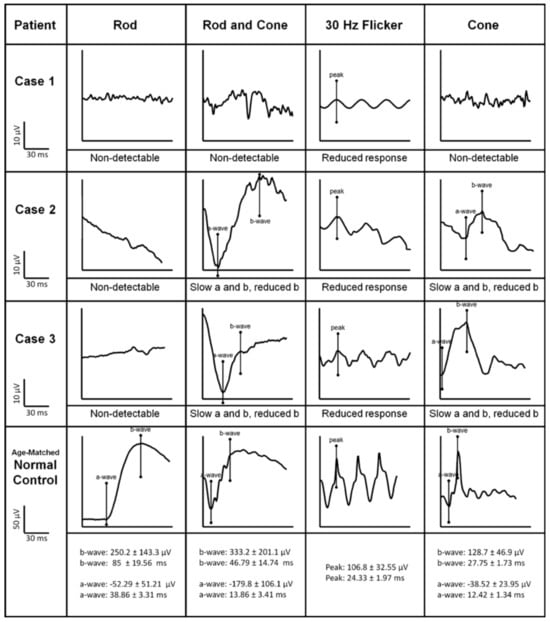
5. Bradyopsia
6. Bietti Crystalline Dystrophy
7. Late-Onset Retinal Degeneration (L-ORD)
8. Fundus Albipunctatus
9. Retinitis Punctata Albescens
References
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40.
- Ebdali, S.; Hashemi, B.; Hashemi, H.; Jafarzadehpur, E.; Asgari, S. Time and frequency components of ERG responses in retinitis pigmentosa. Int. Ophthalmol. 2018, 38, 2435–2444.
- Hassan-Karimi, H.; Jafarzadehpur, E.; Blouri, B.; Hashemi, H.; Sadeghi, A.Z.; Mirzajani, A. Frequency Domain Electroretinography in Retinitis Pigmentosa versus Normal Eyes. J. Ophthalmic Vis. Res. 2012, 7, 34–38.
- Arsiwalla, T.A.; Cornish, E.E.; Nguyen, P.V.; Korsakova, M.; Ali, H.; Saakova, N.; Fraser, C.L.; Jamieson, R.V.; Grigg, J.R. Assessing Residual Cone Function in Retinitis Pigmentosa Patients. Transl. Vis. Sci. Technol. 2020, 9, 29.
- Karali, M.; Testa, F.; Brunetti-Pierri, R.; Di Iorio, V.; Pizzo, M.; Melillo, P.; Barillari, M.R.; Torella, A.; Musacchia, F.; D’Angelo, L.; et al. Clinical and Genetic Analysis of a European Cohort with Pericentral Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 21, 86.
- Huang, C.W.; Yang, J.J.; Yang, C.H.; Yang, C.M.; Hu, F.R.; Ho, T.C.; Chen, T.C. The structure-function correlation analysed by OCT and full field ERG in typical and pericentral subtypes of retinitis pigmentosa. Sci. Rep. 2021, 11, 16883.
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186.
- Todorova, M.G.; Turksever, C.; Schotzau, A.; Schorderet, D.F.; Valmaggia, C. Metabolic and functional changes in retinitis pigmentosa: Comparing retinal vessel oximetry to full-field electroretinography, electrooculogram and multifocal electroretinography. Acta Ophthalmol. 2016, 94, e231–e241.
- Hood, D.C.; Holopigian, K.; Greenstein, V.; Seiple, W.; Li, J.; Sutter, E.E.; Carr, R.E. Assessment of local retinal function in patients with retinitis pigmentosa using the multi-focal ERG technique. Vis. Res. 1998, 38, 163–179.
- Moschos, M.M.; Chatziralli, I.P.; Verriopoulos, G.; Triglianos, A.; Ladas, D.S.; Brouzas, D. Correlation between optical coherence tomography and multifocal electroretinogram findings with visual acuity in retinitis pigmentosa. Clin. Ophthalmol. 2013, 7, 2073–2078.
- Okado, S.; Koyanagi, Y.; Inooka, T.; Kominami, T.; Terasaki, H.; Nishiguchi, K.M.; Ueno, S. Assessments of Macular Function by Focal Macular Electroretinography and Static Perimetry in Eyes with Retinitis Pigmentosa. Retina 2022, 42, 2184–2193.
- Gerth, C.; Wright, T.; Heon, E.; Westall, C.A. Assessment of central retinal function in patients with advanced retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1312–1318.
- Konieczka, K.; Bojinova, R.I.; Valmaggia, C.; Schorderet, D.F.; Todorova, M.G. Preserved functional and structural integrity of the papillomacular area correlates with better visual acuity in retinitis pigmentosa. Eye 2016, 30, 1310–1323.
- Arden, G.B.; Wolf, J.E. The electro-oculographic responses to alcohol and light in a series of patients with retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2730–2734.
- Vingolo, E.M.; Livani, M.L.; Domanico, D.; Mendonca, R.H.; Rispoli, E. Optical coherence tomography and electro-oculogram abnormalities in X-linked retinitis pigmentosa. Doc. Ophthalmol. 2006, 113, 5–10.
- Pinckers, A.; van Aarem, A.; Brink, H. The electrooculogram in heterozygote carriers of Usher syndrome, retinitis pigmentosa, neuronal ceroid lipofuscinosis, senior syndrome and choroideremia. Ophthalmic Genet. 1994, 15, 25–30.
- Asanad, S.; Karanjia, R. Multifocal Electroretinogram; StatPearls Publishing: Treasure Island, FL, USA, 2023.
- Parisi, V.; Ziccardi, L.; Stifano, G.; Montrone, L.; Gallinaro, G.; Falsini, B. Impact of regional retinal responses on cortical visually evoked responses: Multifocal ERGs and VEPs in the retinitis pigmentosa model. Clin. Neurophysiol. 2010, 121, 380–385.
- Granse, L.; Ponjavic, V.; Andreasson, S. Full-field ERG, multifocal ERG and multifocal VEP in patients with retinitis pigmentosa and residual central visual fields. Acta Ophthalmol. Scand. 2004, 82, 701–706.
- Simunovic, M.P.; Moore, A.T. The cone dystrophies. Eye 1998, 12, 553–565.
- Szlyk, J.P.; Fishman, G.A.; Alexander, K.R.; Peachey, N.S.; Derlacki, D.J. Clinical subtypes of cone-rod dystrophy. Arch. Ophthalmol. 1993, 111, 781–788.
- Kamenarova, K.; Corton, M.; Garcia-Sandoval, B.; Fernandez-San Jose, P.; Panchev, V.; Avila-Fernandez, A.; Lopez-Molina, M.I.; Chakarova, C.; Ayuso, C.; Bhattacharya, S.S. Novel GUCA1A mutations suggesting possible mechanisms of pathogenesis in cone, cone-rod, and macular dystrophy patients. BioMed Res. Int. 2013, 2013, 517570.
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7.
- Downes, S.M.; Holder, G.E.; Fitzke, F.W.; Payne, A.M.; Warren, M.J.; Bhattacharya, S.S.; Bird, A.C. Autosomal dominant cone and cone-rod dystrophy with mutations in the guanylate cyclase activator 1A gene-encoding guanylate cyclase activating protein-1. Arch. Ophthalmol. 2001, 119, 96–105.
- Garafalo, A.V.; Sheplock, R.; Sumaroka, A.; Roman, A.J.; Cideciyan, A.V.; Jacobson, S.G. Childhood-onset genetic cone-rod photoreceptor diseases and underlying pathobiology. eBioMedicine 2021, 63, 103200.
- Michaelides, M.; Hardcastle, A.J.; Hunt, D.M.; Moore, A.T. Progressive cone and cone-rod dystrophies: Phenotypes and underlying molecular genetic basis. Surv. Ophthalmol. 2006, 51, 232–258.
- Wang, I.; Khan, N.W.; Branham, K.; Wissinger, B.; Kohl, S.; Heckenlively, J.R. Establishing baseline rod electroretinogram values in achromatopsia and cone dystrophy. Doc. Ophthalmol. 2012, 125, 229–233.
- Zahlava, J.; Lestak, J.; Karel, I. Optical coherence tomography in progressive cone dystrophy. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech Repub. 2014, 158, 628–634.
- Robson, A.G.; Webster, A.R.; Michaelides, M.; Downes, S.M.; Cowing, J.A.; Hunt, D.M.; Moore, A.T.; Holder, G.E. “Cone dystrophy with supernormal rod electroretinogram”: A comprehensive genotype/phenotype study including fundus autofluorescence and extensive electrophysiology. Retina 2010, 30, 51–62.
- Robson, A.G.; Michaelides, M.; Saihan, Z.; Bird, A.C.; Webster, A.R.; Moore, A.T.; Fitzke, F.W.; Holder, G.E. Functional characteristics of patients with retinal dystrophy that manifest abnormal parafoveal annuli of high density fundus autofluorescence; a review and update. Doc. Ophthalmol. 2008, 116, 79–89.
- Vincent, A.; Robson, A.G.; Holder, G.E. Pathognomonic (diagnostic) ERGs. A review and update. Retina 2013, 33, 5–12.
- Hull, S.; Arno, G.; Sergouniotis, P.I.; Tiffin, P.; Borman, A.D.; Chandra, A.; Robson, A.G.; Holder, G.E.; Webster, A.R.; Moore, A.T. Clinical and molecular characterization of enhanced S-cone syndrome in children. JAMA Ophthalmol. 2014, 132, 1341–1349.
- Marmor, M.F.; Jacobson, S.G.; Foerster, M.H.; Kellner, U.; Weleber, R.G. Diagnostic clinical findings of a new syndrome with night blindness, maculopathy, and enhanced S cone sensitivity. Am. J. Ophthalmol. 1990, 110, 124–134.
- Pachydaki, S.I.; Klaver, C.C.; Barbazetto, I.A.; Roy, M.S.; Gouras, P.; Allikmets, R.; Yannuzzi, L.A. Phenotypic features of patients with NR2E3 mutations. Arch. Ophthalmol. 2009, 127, 71–75.
- Milam, A.H.; Rose, L.; Cideciyan, A.V.; Barakat, M.R.; Tang, W.X.; Gupta, N.; Aleman, T.S.; Wright, A.F.; Stone, E.M.; Sheffield, V.C.; et al. The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 473–478.
- Coppieters, F.; Leroy, B.P.; Beysen, D.; Hellemans, J.; De Bosscher, K.; Haegeman, G.; Robberecht, K.; Wuyts, W.; Coucke, P.J.; De Baere, E. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am. J. Hum. Genet. 2007, 81, 147–157.
- Escher, P.; Gouras, P.; Roduit, R.; Tiab, L.; Bolay, S.; Delarive, T.; Chen, S.; Tsai, C.C.; Hayashi, M.; Zernant, J.; et al. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum. Mutat. 2009, 30, 342–351.
- Alsalamah, A.K.; Khan, A.O.; Bakar, A.A.; Schatz, P.; Nowilaty, S.R. Recognizable Patterns of Submacular Fibrosis in Enhanced S-Cone Syndrome. Ophthalmol. Retina 2021, 5, 918–927.
- Sustar, M.; Perovsek, D.; Cima, I.; Stirn-Kranjc, B.; Hawlina, M.; Brecelj, J. Electroretinography and optical coherence tomography reveal abnormal post-photoreceptoral activity and altered retinal lamination in patients with enhanced S-cone syndrome. Doc. Ophthalmol. 2015, 130, 165–177.
- Lam, B.L.; Goldberg, J.L.; Hartley, K.L.; Stone, E.M.; Liu, M. Atypical mild enhanced S-cone syndrome with novel compound heterozygosity of the NR2E3 gene. Am. J. Ophthalmol. 2007, 144, 157–159.
- Naik, A.; Ratra, D.; Banerjee, A.; Dalan, D.; Jandyal, S.; Rao, G.; Sen, P.; Bhende, M.; Jayaprakash, V.; Susvar, P.; et al. Enhanced S-cone syndrome: Clinical spectrum in Indian population. Indian J. Ophthalmol. 2019, 67, 523–529.
- Audo, I.; Michaelides, M.; Robson, A.G.; Hawlina, M.; Vaclavik, V.; Sandbach, J.M.; Neveu, M.M.; Hogg, C.R.; Hunt, D.M.; Moore, A.T.; et al. Phenotypic variation in enhanced S-cone syndrome. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2082–2093.
- De Carvalho, E.R.; Robson, A.G.; Arno, G.; Boon, C.J.F.; Webster, A.A.; Michaelides, M. Enhanced S-Cone Syndrome: Spectrum of Clinical, Imaging, Electrophysiologic, and Genetic Findings in a Retrospective Case Series of 56 Patients. Ophthalmol. Retina 2021, 5, 195–214.
- Park, S.P.; Hong, I.H.; Tsang, S.H.; Lee, W.; Horowitz, J.; Yzer, S.; Allikmets, R.; Chang, S. Disruption of the human cone photoreceptor mosaic from a defect in NR2E3 transcription factor function in young adults. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 2299–2309.
- Michaelides, M.; Li, Z.; Rana, N.A.; Richardson, E.C.; Hykin, P.G.; Moore, A.T.; Holder, G.E.; Webster, A.R. Novel mutations and electrophysiologic findings in RGS9- and R9AP-associated retinal dysfunction (Bradyopsia). Ophthalmology 2010, 117, 120–127.e1.
- Nishiguchi, K.M.; Sandberg, M.A.; Kooijman, A.C.; Martemyanov, K.A.; Pott, J.W.; Hagstrom, S.A.; Arshavsky, V.Y.; Berson, E.L.; Dryja, T.P. Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature 2004, 427, 75–78.
- Lyubarsky, A.L.; Naarendorp, F.; Zhang, X.; Wensel, T.; Simon, M.I.; Pugh, E.N., Jr. RGS9-1 is required for normal inactivation of mouse cone phototransduction. Mol. Vis. 2001, 7, 71–78.
- Chen, C.K.; Burns, M.E.; He, W.; Wensel, T.G.; Baylor, D.A.; Simon, M.I. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9-1. Nature 2000, 403, 557–560.
- Lee, K.Y.; Koh, A.H.; Aung, T.; Yong, V.H.; Yeung, K.; Ang, C.L.; Vithana, E.N. Characterization of Bietti crystalline dystrophy patients with CYP4V2 mutations. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3812–3816.
- Mansour, A.M.; Uwaydat, S.H.; Chan, C.C. Long-term follow-up in Bietti crystalline dystrophy. Eur. J. Ophthalmol. 2007, 17, 680–682.
- Usui, T.; Tanimoto, N.; Takagi, M.; Hasegawa, S.; Abe, H. Rod and cone a-waves in three cases of Bietti crystalline chorioretinal dystrophy. Am. J. Ophthalmol. 2001, 132, 395–402.
- Sen, P.; Ray, R.; Ravi, P. Electrophysiological findings in Bietti’s crystalline dystrophy. Clin. Exp. Optom. 2011, 94, 302–308.
- Saatci, A.O.; Atas, F.; Cetin, G.O.; Kayabasi, M. Diagnostic and Management Strategies of Bietti Crystalline Dystrophy: Current Perspectives. Clin. Ophthalmol. 2023, 17, 953–967.
- Rossi, S.; Testa, F.; Li, A.; Iorio, V.D.; Zhang, J.; Gesualdo, C.; Corte, M.D.; Chan, C.C.; Fielding Hejtmancik, J.; Simonelli, F. An atypical form of Bietti crystalline dystrophy. Ophthalmic Genet. 2011, 32, 118–121.
- Garcia-Garcia, G.P.; Martinez-Rubio, M.; Moya-Moya, M.A.; Perez-Santonja, J.J.; Escribano, J. Current perspectives in Bietti crystalline dystrophy. Clin. Ophthalmol. 2019, 13, 1379–1399.
- Lai, T.Y.; Ng, T.K.; Tam, P.O.; Yam, G.H.; Ngai, J.W.; Chan, W.M.; Liu, D.T.; Lam, D.S.; Pang, C.P. Genotype phenotype analysis of Bietti’s crystalline dystrophy in patients with CYP4V2 mutations. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5212–5220.
- Lockhart, C.M.; Smith, T.B.; Yang, P.; Naidu, M.; Rettie, A.E.; Nath, A.; Weleber, R.; Kelly, E.J. Longitudinal characterisation of function and structure of Bietti crystalline dystrophy: Report on a novel homozygous mutation in CYP4V2. Br. J. Ophthalmol. 2018, 102, 187–194.
- Borooah, S.; Collins, C.; Wright, A.; Dhillon, B. Late-onset retinal macular degeneration: Clinical insights into an inherited retinal degeneration. Br. J. Ophthalmol. 2009, 93, 284–289.
- Ayyagari, R.; Mandal, M.N.; Karoukis, A.J.; Chen, L.; McLaren, N.C.; Lichter, M.; Wong, D.T.; Hitchcock, P.F.; Caruso, R.C.; Moroi, S.E.; et al. Late-onset macular degeneration and long anterior lens zonules result from a CTRP5 gene mutation. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3363–3371.
- Soumplis, V.; Sergouniotis, P.I.; Robson, A.G.; Michaelides, M.; Moore, A.T.; Holder, G.E.; Webster, A.R. Phenotypic findings in C1QTNF5 retinopathy (late-onset retinal degeneration). Acta Ophthalmol. 2013, 91, e191–e195.
- Milam, A.H.; Curcio, C.A.; Cideciyan, A.V.; Saxena, S.; John, S.K.; Kruth, H.S.; Malek, G.; Heckenlively, J.R.; Weleber, R.G.; Jacobson, S.G. Dominant late-onset retinal degeneration with regional variation of sub-retinal pigment epithelium deposits, retinal function, and photoreceptor degeneration. Ophthalmology 2000, 107, 2256–2266.
- Vincent, A.; Munier, F.L.; Vandenhoven, C.C.; Wright, T.; Westall, C.A.; Heon, E. The characterization of retinal phenotype in a family with C1QTNF5-related late-onset retinal degeneration. Retina 2012, 32, 1643–1651.
- Kuntz, C.A.; Jacobson, S.G.; Cideciyan, A.V.; Li, Z.Y.; Stone, E.M.; Possin, D.; Milam, A.H. Sub-retinal pigment epithelial deposits in a dominant late-onset retinal degeneration. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1772–1782.
- De Zaeytijd, J.; Coppieters, F.; De Bruyne, M.; Van Royen, J.; Roels, D.; Six, R.; Van Cauwenbergh, C.; De Baere, E.; Leroy, B.P. Longitudinal phenotypic study of late-onset retinal degeneration due to a founder variant c.562C>A p.(Pro188Thr) in the C1QTNF5 gene. Ophthalmic Genet. 2021, 42, 521–532.
- Papastavrou, V.T.; Bradshaw, K.R.; Aye, K.H.; Turney, C.; Browning, A.C. Improvement of retinal function in L-ORD after prolonged dark adaptation. Can. J. Ophthalmol. 2015, 50, 112–118.
- Borooah, S.; Papastavrou, V.; Lando, L.; Han, J.; Lin, J.H.; Ayyagari, R.; Dhillon, B.; Browning, A.C. Reticular Pseudodrusen in Late-Onset Retinal Degeneration. Ophthalmol. Retina 2021, 5, 1043–1051.
- Lando, L.; Borooah, S. Late-Onset Retinal Degeneration: Clinical Perspectives. Clin. Ophthalmol. 2022, 16, 3225–3246.
- Nakamura, M.; Hotta, Y.; Tanikawa, A.; Terasaki, H.; Miyake, Y. A high association with cone dystrophy in Fundus albipunctatus caused by mutations of the RDH5 gene. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3925–3932.
- Sergouniotis, P.I.; Sohn, E.H.; Li, Z.; McBain, V.A.; Wright, G.A.; Moore, A.T.; Robson, A.G.; Holder, G.E.; Webster, A.R. Phenotypic variability in RDH5 retinopathy (Fundus Albipunctatus). Ophthalmology 2011, 118, 1661–1670.
- Nakamura, M.; Skalet, J.; Miyake, Y. RDH5 gene mutations and electroretinogram in fundus albipunctatus with or without macular dystrophy: RDH5 mutations and ERG in fundus albipunctatus. Doc. Ophthalmol. 2003, 107, 3–11.
- Hajali, M.; Fishman, G.A.; Dryja, T.P.; Sweeney, M.O.; Lindeman, M. Diagnosis in a patient with fundus albipunctatus and atypical fundus changes. Doc. Ophthalmol. 2009, 118, 233–238.
- Fishman, G.A.; Roberts, M.F.; Derlacki, D.J.; Grimsby, J.L.; Yamamoto, H.; Sharon, D.; Nishiguchi, K.M.; Dryja, T.P. Novel mutations in the cellular retinaldehyde-binding protein gene (RLBP1) associated with retinitis punctata albescens: Evidence of interfamilial genetic heterogeneity and fundus changes in heterozygotes. Arch. Ophthalmol. 2004, 122, 70–75.
- Granse, L.; Abrahamson, M.; Ponjavic, V.; Andreasson, S. Electrophysiological findings in two young patients with Bothnia dystrophy and a mutation in the RLBP1 gene. Ophthalmic Genet. 2001, 22, 97–105.

