 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dhiren Patel | -- | 2890 | 2023-11-16 17:01:08 | | | |
| 2 | Sirius Huang | Meta information modification | 2890 | 2023-11-17 09:21:57 | | |
Video Upload Options
Cystic Fibrosis is a chronic disease affecting multiple systems, including the GI tract. Clinical manifestation in patients can start as early as infancy and vary across different age groups. With the advent of new, highly effective modulators, the life expectancy of people with CF (PwCF) has improved significantly. Various GI aspects of CF care, such as nutrition, are linked to an overall improvement in morbidity, lung function and the quality of life of PwCF. The variable clinical presentations and management of GI diseases in pediatrics and adults with CF should be recognized. Therefore, it is necessary to ensure efficient transfer of information between pediatric and adult providers for proper continuity of management and coordination of care at the time of transition.
1. Introduction
2. Esophagus and Stomach
2.1. GERD and Foregut Dysmotility
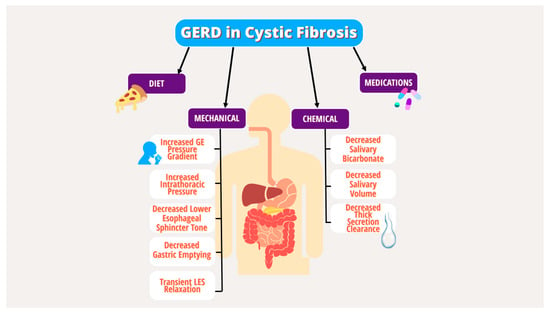
Pediatrics
Adults
Transition Take-Home Points
- Reassess the indication for PPI and consider discontinuation trial.
- Screen for alarm features or poor response to therapy and consider upper endoscopy and surgical referral when clinically appropriate due to a higher risk of Barrett’s esophagus and esophageal cancer.
3. Intestine
3.1. Small Intestinal Bacterial Overgrowth
Pediatrics
Adults
3.2. Intestinal Obstruction Syndromes: Meconium Ileus, DIOS, Constipation
Pediatrics
Adults
Transition Take-Home Points
- Identify patients with nutritional deficiencies with symptoms of SIBO and consider for diagnostic evaluation or empiric therapy.
- Identify patients with a history of meconium ileus requiring surgical intervention during childhood or PI who are at higher risk of DIOS and constipation.
- Obtain a thorough history of previously tolerated and unsuccessful treatments for constipation to guide future care which can include optimizing PERT dosing, adjusting bowel regimens, and consideration of combination therapies.
References
- Scotet, V.; L’Hostis, C.; Férec, C. The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes 2020, 11, 589.
- McBennett, K.A.; Davis, P.B.; Konstan, M.W. Increasing life expectancy in cystic fibrosis: Advances and challenges. Pediatr. Pulmonol. 2022, 57 (Suppl. S1), S5–S12.
- Tuchman, L.K.; Schwartz, L.A.; Sawicki, G.S.; Britto, M.T. Cystic fibrosis and transition to adult medical care. Pediatrics 2010, 125, 566–573.
- Goralski, J.L.; Nasr, S.Z.; Uluer, A. Overcoming barriers to a successful transition from pediatric to adult care. Pediatr. Pulmonol. 2017, 52, S52–S60.
- Transition, G. Got Transition. The Six Core Elements of Health Care Transition. Available online: http://www.gottransition.org/providers/index.cfm (accessed on 15 September 2022).
- Baker, A.M.; Riekert, K.A.; Sawicki, G.S.; Eakin, M.N. CF RISE: Implementing a Clinic-Based Transition Program. Pediatr. Allergy Immunol. Pulmonol. 2015, 28, 250–254.
- Van Biervliet, S.; de Clercq, C.; Declercq, D.; Van Braeckel, E.; Van Daele, S.; De Baets, F.; De Looze, D. Gastro-intestinal manifestations in cystic fibrosis patients. Acta Gastroenterol. Belg. 2016, 79, 481–486.
- Gustafsson, P.M.; Fransson, S.G.; Kjellman, N.I.; Tibbling, L. Gastro-oesophageal reflux and severity of pulmonary disease in cystic fibrosis. Scand. J. Gastroenterol. 1991, 26, 449–456.
- Palm, K.; Sawicki, G.; Rosen, R. The impact of reflux burden on Pseudomonas positivity in children with Cystic Fibrosis. Pediatric. Pulmonol. 2012, 47, 582–587.
- Pauwels, A.; Blondeau, K.; Dupont, L.J.; Sifrim, D. Mechanisms of increased gastroesophageal reflux in patients with cystic fibrosis. Am. J. Gastroenterol. 2012, 107, 1346–1353.
- Stringer, D.A.; Sprigg, A.; Juodis, E.; Corey, M.; Daneman, A.; Levison, H.J.; Durie, P.R. The association of cystic fibrosis, gastroesophageal reflux, and reduced pulmonary function. Can. Assoc. Radiol. J. 1988, 39, 100–102.
- Ng, C.; Prayle, A.P. Gastrointestinal complications of cystic fibrosis. Paediatr. Child Health 2020, 30, 345–349.
- Assis, D.N.; Freedman, S.D. Gastrointestinal Disorders in Cystic Fibrosis. Clin. Chest Med. 2016, 37, 109–118.
- Maqbool, A.; Pauwels, A. Cystic Fibrosis and gastroesophageal reflux disease. J. Cyst. Fibros. 2017, 16 (Suppl. S2), S2–S13.
- Lewindon, P.J.; Robb, T.A.; Moore, D.J.; Davidson, G.P.; Martin, A.J. Bowel dysfunction in cystic fibrosis: Importance of breath testing. J. Paediatr. Child Health 1998, 34, 79–82.
- Mousa, H.M.; Woodley, F.W. Gastroesophageal reflux in cystic fibrosis: Current understandings of mechanisms and management. Curr. Gastroenterol. Rep. 2012, 14, 226–235.
- Bongiovanni, A.; Manti, S.; Parisi, G.F.; Papale, M.; Mulè, E.; Rotolo, N.; Leonardi, S. Focus on gastroesophageal reflux disease in patients with cystic fibrosis. World J. Gastroenterol. 2020, 26, 6322–6334.
- Henen, S.; Denton, C.; Teckman, J.; Borowitz, D.; Patel, D. Review of Gastrointestinal Motility in Cystic Fibrosis. J. Cyst. Fibros. 2021, 20, 578–585.
- Gelfond, D.; Ma, C.; Semler, J.; Borowitz, D. Intestinal pH and gastrointestinal transit profiles in cystic fibrosis patients measured by wireless motility capsule. Dig. Dis. Sci. 2013, 58, 2275–2281.
- Pauwels, A.; Decraene, A.; Blondeau, K.; Mertens, V.; Farre, R.; Proesmans, M.; Van Bleyenbergh, P.; Sifrim, D.; Dupont, L.J. Bile acids in sputum and increased airway inflammation in patients with cystic fibrosis. Chest 2012, 141, 1568–1574.
- Hedsund, C.; Gregersen, T.; Joensson, I.M.; Olesen, H.V.; Krogh, K. Gastrointestinal transit times and motility in patients with cystic fibrosis. Scand. J. Gastroenterol. 2012, 47, 920–926.
- Corral, J.E.; Dye, C.W.; Mascarenhas, M.R.; Barkin, J.S.; Salathe, M.; Moshiree, B. Is Gastroparesis Found More Frequently in Patients with Cystic Fibrosis? A Systematic Review. Scientifica 2016, 2016, 2918139.
- Vandenplas, Y.; Rudolph, C.D.; Di Lorenzo, C.; Hassall, E.; Liptak, G.; Mazur, L.; Sondheimer, J.; Staiano, A.; Thomson, M.; Veereman-Wauters, G.; et al. Pediatric gastroesophageal reflux clinical practice guidelines: Joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition (NASPGHAN) and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN). J. Pediatr. Gastroenterol. Nutr. 2009, 49, 498–547.
- Mertens, V.; Blondeau, K.; Pauwels, A.; Farre, R.; Vanaudenaerde, B.; Vos, R.; Verleden, G.; Van Raemdonck, D.E.; Dupont, L.J.; Sifrim, D. Azithromycin reduces gastroesophageal reflux and aspiration in lung transplant recipients. Dig. Dis. Sci. 2009, 54, 972–979.
- Boesch, R.P.; Acton, J.D. Outcomes of fundoplication in children with cystic fibrosis. J. Pediatr. Surg. 2007, 42, 1341–1344.
- Sheikh, S.I.; Ryan-Wenger, N.A.; McCoy, K.S. Outcomes of surgical management of severe GERD in patients with cystic fibrosis. Pediatr. Pulmonol. 2013, 48, 556–562.
- Blondeau, K.; Dupont, L.J.; Mertens, V.; Verleden, G.; Malfroot, A.; Vandenplas, Y.; Hauser, B.; Sifrim, D. Gastro-oesophageal reflux and aspiration of gastric contents in adult patients with cystic fibrosis. Gut 2008, 57, 1049–1055.
- Lusman, S.S.; Grand, R. Approach to chronic abdominal pain in Cystic Fibrosis. J. Cyst. Fibros. 2017, 16 (Suppl. S2), S24–S31.
- Knotts, R.M.; Solfisburg, Q.S.; Keating, C.; DiMango, E.; Lightdale, C.J.; Abrams, J.A. Cystic fibrosis is associated with an increased risk of Barrett’s esophagus. J. Cyst. Fibros. 2019, 18, 425–429.
- Fridge, J.L.; Conrad, C.; Gerson, L.; Castillo, R.O.; Cox, K. Risk factors for small bowel bacterial overgrowth in cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 2007, 44, 212–218.
- Lisowska, A.; Wójtowicz, J.; Walkowiak, J. Small intestine bacterial overgrowth is frequent in cystic fibrosis: Combined hydrogen and methane measurements are required for its detection. Acta Biochim. Pol. 2009, 56, 631–634.
- Dorsey, J.; Gonska, T. Bacterial overgrowth, dysbiosis, inflammation, and dysmotility in the Cystic Fibrosis intestine. J. Cyst. Fibros. 2017, 16 (Suppl. S2), S14–S23.
- Malik, B.A.; Xie, Y.Y.; Wine, E.; Huynh, H.Q. Diagnosis and pharmacological management of small intestinal bacterial overgrowth in children with intestinal failure. Can. J. Gastroenterol. 2011, 25, 41–45.
- Haller, W.; Ledder, O.; Lewindon, P.J.; Couper, R.; Gaskin, K.J.; Oliver, M. Cystic fibrosis: An update for clinicians. Part 1: Nutrition and gastrointestinal complications. J. Gastroenterol. Hepatol. 2014, 29, 1344–1355.
- De Lisle, R.C.; Roach, E.; Jansson, K. Effects of laxative and N-acetylcysteine on mucus accumulation, bacterial load, transit, and inflammation in the cystic fibrosis mouse small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G577–G584.
- Quigley, E.M.; Quera, R. Small intestinal bacterial overgrowth: Roles of antibiotics, prebiotics, and probiotics. Gastroenterology 2006, 130, S78–S90.
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280.
- Shah, S.C.; Day, L.W.; Somsouk, M.; Sewell, J.L. Meta-analysis: Antibiotic therapy for small intestinal bacterial overgrowth. Aliment. Pharmacol. Ther. 2013, 38, 925–934.
- Furnari, M.; De Alessandri, A.; Cresta, F.; Haupt, M.; Bassi, M.; Calvi, A.; Haupt, R.; Bodini, G.; Ahmed, I.; Bagnasco, F.; et al. The role of small intestinal bacterial overgrowth in cystic fibrosis: A randomized case-controlled clinical trial with rifaximin. J. Gastroenterol. 2019, 54, 261–270.
- Bali, A.; Stableforth, D.E.; Asquith, P. Prolonged small-intestinal transit time in cystic fibrosis. Br. Med. J. (Clin. Res. Ed.) 1983, 287, 1011–1013.
- Escobar, H.; Perdomo, M.; Vasconez, F.; Camarero, C.; del Olmo, M.T.; Suárez, L. Intestinal permeability to 51Cr-EDTA and orocecal transit time in cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 1992, 14, 204–207.
- Report, A.D. Annual Data Report 2020 Cystic Fibrosis Foundation Patient Registry. Available online: https://www.cff.org/sites/default/files/2021-10/2019-Patient-Registry-Annual-Data-Report.pdf (accessed on 15 September 2022).
- Houwen, R.H.; van der Doef, H.P.; Sermet, I.; Munck, A.; Hauser, B.; Walkowiak, J.; Robberecht, E.; Colombo, C.; Sinaasappel, M.; Wilschanski, M. Defining DIOS and constipation in cystic fibrosis with a multicentre study on the incidence, characteristics, and treatment of DIOS. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 38–42.
- Munck, A.; Alberti, C.; Colombo, C.; Kashirskaya, N.; Ellemunter, H.; Fotoulaki, M.; Houwen, R.; Robberecht, E.; Boizeau, P.; Wilschanski, M. International prospective study of distal intestinal obstruction syndrome in cystic fibrosis: Associated factors and outcome. J. Cyst. Fibros. 2016, 15, 531–539.
- Colombo, C.; Ellemunter, H.; Houwen, R.; Munck, A.; Taylor, C.; Wilschanski, M. Guidelines for the diagnosis and management of distal intestinal obstruction syndrome in cystic fibrosis patients. J. Cyst. Fibros. 2011, 10 (Suppl. S2), S24–S28.
- Declercq, D.; Van Biervliet, S.; Robberecht, E. Nutrition and pancreatic enzyme intake in patients with cystic fibrosis with distal intestinal obstruction syndrome. Nutr. Clin. Pract. 2015, 30, 134–137.
- Andersen, H.O.; Hjelt, K.; Waever, E.; Overgaard, K. The age-related incidence of meconium ileus equivalent in a cystic fibrosis population: The impact of high-energy intake. J. Pediatr. Gastroenterol. Nutr. 1990, 11, 356–360.
- Speck, K.; Charles, A. Distal intestinal obstructive syndrome in adults with cystic fibrosis: A surgical perspective. Arch. Surg. 2008, 143, 601–603.
- O’Brien, C.E.; Anderson, P.J.; Stowe, C.D. Lubiprostone for constipation in adults with cystic fibrosis: A pilot study. Ann. Pharmacother. 2011, 45, 1061–1066.
- Tabbers, M.M.; DiLorenzo, C.; Berger, M.Y.; Faure, C.; Langendam, M.W.; Nurko, S.; Staiano, A.; Vandenplas, Y.; Benninga, M.A. Evaluation and treatment of functional constipation in infants and children: Evidence-based recommendations from ESPGHAN and NASPGHAN. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 258–274.
- O’Brien, C.E.; Anderson, P.J.; Stowe, C.D. Use of the chloride channel activator lubiprostone for constipation in adults with cystic fibrosis: A case series. Ann. Pharmacother. 2010, 44, 577–581.
- McHugh, D.R.; Cotton, C.U.; Moss, F.J.; Vitko, M.; Valerio, D.M.; Kelley, T.J.; Hao, S.; Jafri, A.; Drumm, M.L.; Boron, W.F.; et al. Linaclotide improves gastrointestinal transit in cystic fibrosis mice by inhibiting sodium/hydrogen exchanger 3. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G868–G878.

