1. Introduction
Improved understanding of Cystic Fibrosis (CF) and advances in life-extending treatment over the years have led to considerable changes in the epidemiological profile. While CF was previously considered a lethal pediatric genetic condition, people with CF (PwCF) are now surviving well into adulthood. With increased life expectancy, PwCF face new challenges of living as adults with adult disease manifestations [
1]. This improvement in median predicted survival to approximately 50 years of age has introduced new therapeutic challenges and underscored the need for coordinated, uninterrupted, and developmentally appropriate transitions from pediatric to adult-oriented healthcare [
2]. Navigating this transition can be highly complex for both patients and providers and carries numerous challenges related to patient readiness, poor insight into this complex disease, psychosocial factors (including mental health issues and adverse social circumstances), changes in disease phenotypes with age, and treatment variations [
3,
4]. General tools are available to guide patients and providers in constructing a structured transition program (such as “Got Transition” to help guide care coordination improvement and “CF R.I.S.E” as an objective measure of patient readiness); however, no standardized processes currently exist, and current tools are not specific to GI transitions of care [
5,
6].
CF care requires the involvement of various specialists who play vital roles in transitions of care. The careful collaboration between multidisciplinary CF pediatric and adult teams is therefore imperative to ensure a successful transition, avoid gaps in care, and optimize outcomes. Gastrointestinal (GI) manifestations in particular contribute substantially to extra-pulmonary morbidity in CF and represent a major target for efforts to enhance care transitions [
7]. GI manifestations can be directly related to the underlying disease process or a consequence of treatment. Additionally, GI manifestations can impact disease outcomes through their effects on nutrition, pulmonary function, and overall patient wellness. Unfortunately, there is limited awareness of CF disease presentations and management in general adult medicine and an increasing need for pediatric and adult gastroenterologists who focus on the unique GI manifestations of CF. The recognition and management of complex GI pathology in CF requires coordinated expertise given the frequently atypical disease presentations and variable management approaches. The Cystic Fibrosis Foundation (CFF) has developed multiple tools over the years to tackle these issues. These include the Program for Adult Care Excellence (PACE) awards, the Learning and Leadership Collaboratives (LLC), and the Developing Innovative Gastroenterology Specialty Training (DIGEST) Program [
4]. Through the DIGEST initiative, the CFF offers training awards to support both pediatric and adult gastroenterologists interested in advancing the care of PwCF. However, there is currently no standardized management and care plan for GI issues in PwCF as they transition into adulthood.
2. Esophagus and Stomach
2.1. GERD and Foregut Dysmotility
Gastrointestinal reflux disease (GERD) is a common extrapulmonary manifestation in CF. Several studies have suggested that GERD in CF leads to more severe pulmonary manifestations, though the interplay is complex [
8,
9,
10,
11,
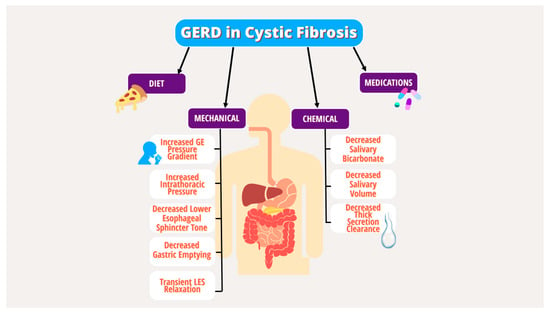
12]. Multiple underlying mechanical and chemical factors have been implicated in the pathogenesis of GERD in PwCF, including increased transient lower esophageal sphincter (LES) relaxation, increased intra-abdominal pressure from coughing, delayed gastric emptying, decreased basal LES tone, lung hyperinflation effects on the diaphragm, and medication effects (
Figure 1) [
10,
13,
14,
15,
16]. Given the lack of specific guidelines for GERD in CF, the diagnostic and therapeutic strategy for GERD in PwCF does not differ from the general population and includes behavioral modifications, dietary changes, medications, and procedures targeting acid production [
17].
Figure 1. Mechanical and chemical factors in the pathogenesis of GERD in PwCF.
GI dysmotility is likely a key factor in the GI manifestations of CF with complex pathophysiology and symptoms that overlap with GERD and dyspepsia. Disruptions in motility are thought to arise from defective epithelial CFTR in the neuromuscular complex acting as a primary insult and leading to altered intestinal myenteric ganglia and abnormal ileal smooth muscle function. Secondary causes of GI dysmotility include decreased bicarbonate and mucoviscidosis from pancreatic insufficiency (PI), fat malabsorption due to poor micelle formation, gut inflammation due to dysbiosis, and hormonal dysregulation [
18]. This is further exacerbated by frequent use of anti-cholinergic medications and opiates. Wireless motility capsules have been used to study GI transit profiles in CF and demonstrated significant delay in small bowel transit time without a compensatory increase in whole gut transit time and deficient buffering capacity required to neutralize gastric acid in the proximal small bowel [
18,
19]. Gastroparesis is a common dysmotility diagnosis in CF that can lead to reduced oral intake, nutritional deficiencies, low body mass index, poor quality of life, and variable response to pancreatic enzyme replacement therapy (PERT). The use of gastric emptying scintigraphy in CF is an area of debate due to inconsistent findings and lack of reproducibility [
18]. Specific guidelines for the management of gastroparesis in CF have not been established.
Pediatrics
In children with CF (CwCF), GERD is the most common GI manifestation. GERD in children can be exacerbated by the risk factors in
Figure 1 and by excessive coughing and use of vest therapy for pulmonary toileting [
10]. About two-thirds of children are asymptomatic, while others manifest symptoms of heartburn, abdominal pain and vomiting. Refusal to eat can be a subtle sign of underlying GERD. Higher incidence of aspiration pneumonia is noted in children with symptomatic GERD [
20]. It is essential to treat GERD effectively, given the possible association with poor pulmonary outcomes [
21]. Unrecognized GERD can limit caloric intake contributing to malnutrition particularly in children, which could be significant for CwCF with higher caloric needs. GERD is a clinical diagnosis, although pH Multichannel Intraluminal Impedance (MII) studies could provide objective evidence. CwCF are on high-dose NSAIDs for anti-inflammatory and protective effects on the lungs, which can adversely cause NSAID-induced gastropathy (gastritis, peptic ulcer disease). Hence, in patients with persistent symptoms despite proton pump inhibitors (PPI) therapy or red flags such as hematemesis, poor growth, esophagogastroduodenoscopy (EGD) should be performed to evaluate for mucosal pathology. Dysmotility in CF affects various portions of GI tract, leading to reflux, dyspepsia, gastroparesis and constipation and can also contribute to refractory reflux symptoms [
21]. A systematic review showed a pooled prevalence of gastroparesis of 38% in CwCF, higher than the general pediatric population [
22].
Lifestyle modifications such as a healthy diet, avoiding spicy food, limiting carbonated/sugary/caffeinated beverages are emphasized. Behavioral changes, such as avoiding head-down postural drainage in infants, can also help to minimize reflux and aspiration. Commonly used acid reducing medications, in children are histamine receptor 2 antagonists (H2-RA) and PPI. Most children remain on acid suppressive therapy long-term, with attempts to wean if asymptomatic and meeting appropriate growth parameters. PPIs are considered to improve efficacy of PERT and used as an adjuvant in management of PI although its true efficacy in modern PERT use is unknown. Prokinetic agents such as metoclopramide, domperidone, and macrolide antibiotics have low efficacy in treating reflux but are commonly used in treating gastroparesis in children [
23]. Azithromycin, a potent anti-inflammatory and pro-kinetic agent, is used to improve gastric motility, although studies have shown it reduces GERD and bile aspiration (specifically after transplantation) in a small subset of CF [
24]. There are controversial data regarding efficacy of fundoplication in CwCF [
25,
26], although it may be considered in medically refractory GERD with comorbidities such as frequent pulmonary exacerbation, recurrent aspiration pneumonia and malnutrition. Apart from reflux esophagitis, other complications such as Barrett’s esophagus and esophageal cancers are rarely seen in the pediatric population.
Adults
In adults with CF (AwCF), there is an increased risk of GERD with an estimated prevalence of 55–90% [
27]. Like CwCF, regurgitation is a common symptom of GERD in AwCF. On the other hand, heartburn is more commonly reported in adolescents and adults with CF. Abdominal pain or dyspepsia in adults can signify the presence of other gastroduodenal pathology such as gastritis, duodenitis, or peptic ulcer disease, which may warrant further evaluation. Dysphagia could be a sign of esophageal dysmotility or esophageal strictures. Concurrent foregut dysmotility or gastroparesis can manifest with post-prandial early satiety, bloating, nausea, and vomiting and can be difficult to clinically distinguish from GERD. Adults with CF on long-term steroids or immunosuppression following transplant are at an increased risk of infectious esophagitis, which may manifest with odynophagia [
28].
As with CwCF, GERD in adults with CF is usually a clinical diagnosis if alarm features (i.e., dysphagia, anemia, weight loss) are absent, and routine management involves dietary and lifestyle modifications and pharmacological acid suppression with PPIs or H2-RAs. Like CwCF, adults with suspected GERD rarely undergo endoscopy for the diagnosis of GERD. Long-term PPI therapy may not always be necessary in AwCF. Thus, during transitions of care, reassessing the indication for PPI use and considering trials of PPI weaning or discontinuation can minimize unnecessary long-term use.
Routine screening for failure to respond to PPI or for the presence of alarm symptoms should be performed at the time of transition. This is especially important considering the substantially increased rate of esophageal adenocarcinoma and Barrett’s esophagus in AwCF compared to the general population with an earlier average age of onset [
29]. Individuals on long-term PPI with inadequate symptom control, intolerance to medications, alarm features, or complications such as Barrett’s esophagus or esophageal strictures should be carefully considered for upper endoscopy and for surgical management if clinically appropriate. In patients with dyspepsia or suspected peptic ulcer disease who are at high risk for endoscopy, non-invasive testing for Helicobacter pylori such as stool antigen test or breath test may also be considered. Due to frequent symptom overlap and despite lack of clear guidelines for the work-up and management of dyspepsia and dysmotility in AwCF, further endoscopic or motility testing can be considered in those who fail to respond to PPI. Though data are lacking, a systematic review of the literature noted an increasing frequency of gastroparesis in CF with increasing age [
22]. This is thought to be due to the increased risk of diabetes in AwCF. The optimal diagnostic approach for gastroparesis in CF remains an area of debate; however, scintigraphy can be considered. If gastroparesis is confirmed, the use of prokinetic agents such as metoclopramide and macrolide antibiotics can be considered as with CwCF. Decompressive gastrostomy tube and gastrojejunostomy feeding tube placement can be considered in cases refractory to medical therapy.
Transition Take-Home Points
- Reassess the indication for PPI and consider discontinuation trial.
- Screen for alarm features or poor response to therapy and consider upper endoscopy and surgical referral when clinically appropriate due to a higher risk of Barrett’s esophagus and esophageal cancer.
3. Intestine
3.1. Small Intestinal Bacterial Overgrowth
There is a well-established correlation between small intestinal bacterial overgrowth (SIBO) and CF in both children and adults. In both populations, establishing a diagnosis can be challenging. The prevalence of SIBO in individuals with CF has been documented to be up to 56% [
15,
30,
31]. The proposed mechanism of SIBO in CF is attributed to a combination of dysmotility, altered surgical anatomy, frequent antibiotic use, and the accumulation of thick mucus secretions in the small bowel acting as a bacterial anchor. Symptoms in children and adults are similar and can include diarrhea, abdominal pain, flatulence, and weight loss. SIBO in CF may carry nutritional implications related to malabsorption and symptom severity.
Pediatrics
SIBO has been reported in almost half of PwCF [
31]. Although duodenal aspirate (bacterial count >10
6 colony forming units/mL and culture) is the gold standard, it is rarely conducted in children. Non-invasive testing, like the hydrogen breath test, is not reliable and difficult to perform in younger children. Diagnosis of SIBO in CwCF is mostly based on clinical symptoms [
30,
32,
33]. In children, symptoms of SIBO can be hard to differentiate from symptoms of malabsorption. If children have prolonged diarrhea, flatulence, poor weight gain despite good caloric intake and optimal PERT, SIBO should be considered [
34]. Treatment includes a trial of oral antibiotics. Osmotic laxatives can be beneficial in treating SIBO by preventing stasis and increasing bacterial excretion [
12,
35]. Prebiotics and probiotics are safe and effective in CwCF [
36]. Efficacy of other medications such as N- acetyl cystine and inhaled ipratropium are still under research [
30,
35].
Adults
Adults with CF are at higher risk for recurrent SIBO due to the longer disease duration and recurrent antibiotics exposure, which alters the gut microbiota over time [
37]. In AwCF, symptoms include bloating, flatulence, diarrhea, weight loss and malnutrition. Nutritional deficiencies in SIBO include fat-soluble vitamins, iron, vitamin b12. The diagnostic methods are similar to what have been reviewed in children. Empiric treatment of SIBO with rifaximin has also been proven to be effective, well tolerated and currently is the gold standard treatment in both adults and pediatrics, although lack of reliable health insurance coverage can lead to treatment delay. [
38,
39]. During care transitions, PwCF with nutritional deficiencies or symptoms of SIBO should be considered for diagnostic evaluation or empiric therapy.
3.2. Intestinal Obstruction Syndromes: Meconium Ileus, DIOS, Constipation
Intestinal obstructions syndromes are interrelated and include meconium ileus at birth, distal intestinal obstruction syndrome (DIOS), and constipation. They occur mainly due to increased or decreased transit time, PI, and the high mucus viscosity in the intestinal tract which leads to intestinal obstruction [
40,
41]. Variable treatment options are utilized but none have good supporting evidence. More randomized controlled trials are needed.
Pediatrics
Around 15% of infants with CF are diagnosed with meconium ileus (MI), which commonly presents in the neonatal period when inspissated meconium causes obstruction. [
42]. Simple MI is treated with hyperosmolar enema (gastrografin, N- acetylcysteine enema), while complicated MI (with perforation, peritonitis, volvulus) needs surgical intervention. During transitions of care, it is important to highlight if the patient has a history of MI and if it required surgical intervention due to potential complications and increased risk for DIOS.
DIOS is a unique condition in CF that presents as acute, complete or partial fecal obstruction, more commonly in the ileocecal region. Patients with severe CFTR mutation, history of meconium ileus, PI, history of prior DIOS and CFRD are at higher risk for DIOS [
43,
44]. Adequate management of chronic constipation and optimization of PERT has been noted to prevent DIOS [
45,
46,
47]. Dietary avoidance of bulky high fiber foods should be advised. Medical management includes IV fluids, laxatives (polyethylene glycol) and gastrografin enemas. Surgery is recommended for failure of conservative management or complications like perforation, peritonitis, or sepsis and should be avoided if possible [
41].
About 50% of CwCF have chronic constipation [
44], due to risk factors such as altered intraluminal fluid-ion composition, dysmotility, and dysbiosis. Constipation has a gradual symptom onset compared to DIOS, which presents acutely. Although there is lack of evidence, exercise and adequate water intake are emphasized. Daily laxative regimens consisting of osmotic laxatives (preferably PEG) are advised. Stimulant laxatives like senna and bisacodyl are considered second line. Failure to maintain a daily laxative regimen and early signs of DIOS should prompt an increased dose of osmotic/stimulant laxatives. Enemas may be utilized to avoid complications and the need for hospitalization.
Adults
While meconium ileus is an exclusively pediatric condition in CF, DIOS, formally known as a meconium ileus equivalent, is more commonly seen in AwCF with a peak incidence between 20–25 years of age [
47]. The definition was specified by the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) to make a clear distinction between DIOS and constipation [
43]. DIOS has varying presentations ranging from incomplete obstruction to acute complete obstruction. Treatment is mainly conservative with fluids, nasogastric decompression, and enemas [
45]. Surgical intervention may be warranted with failure to respond to conservative management or with signs of perforation or ischemia [
48].
As with pediatrics, constipation is one of the more common GI symptoms in CF, and prevalence increases with age. Compared to DIOS, constipation has a more gradual and subtle presentation but it can be challenging to differentiate from DIOS [
49]. Osmotic laxatives are the initial treatment for AwCF with constipation. Stimulant laxatives can be used as an additional treatment [
50]. If the initial treatment fails, other options include Lubiprostone (chloride channel activator) and Linaclotide (guanylate cyclase-C agonist) [
49,
51,
52]. Limited data exist on the best option, as few have been studied in PwCF.
During transitions of care, providers should identify individuals at risk of DIOS including those with prior surgical intervention for meconium ileus or PI. For individuals with a history of constipation, obtaining a thorough history of previously tolerated and unsuccessful treatments can guide future care. Optimizing PERT dosing, adjusting bowel regimens and adding chloride channel activators or other bowel regimens when indicated can help prevent DIOS and relieve constipation.
Transition Take-Home Points
- Identify patients with nutritional deficiencies with symptoms of SIBO and consider for diagnostic evaluation or empiric therapy.
- Identify patients with a history of meconium ileus requiring surgical intervention during childhood or PI who are at higher risk of DIOS and constipation.
- Obtain a thorough history of previously tolerated and unsuccessful treatments for constipation to guide future care which can include optimizing PERT dosing, adjusting bowel regimens, and consideration of combination therapies.
This entry is adapted from the peer-reviewed paper 10.3390/ijms242115766