 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ahilanandan Dushianthan | -- | 2601 | 2023-11-16 11:58:37 | | | |
| 2 | Wendy Huang | Meta information modification | 2601 | 2023-11-16 12:14:04 | | | | |
| 3 | Wendy Huang | Meta information modification | 2601 | 2023-11-20 10:07:30 | | |
Video Upload Options
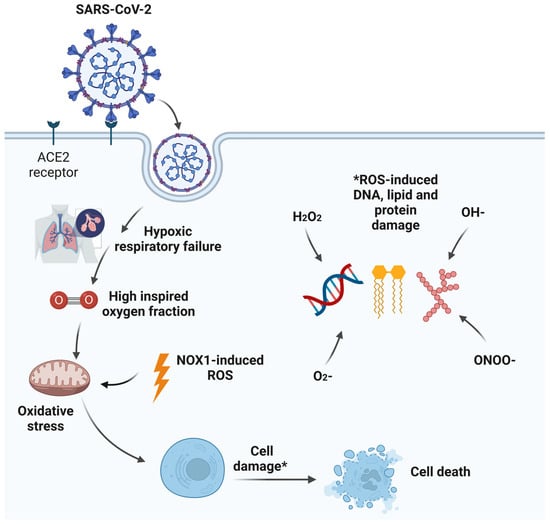
Acute hypoxic respiratory failure (AHRF) is a prominent feature of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) critical illness. The need for a high FiO2 to normalise arterial hypoxemia and tissue hypoxia can result in alveolar hyperoxia. This in turn can lead to local alveolar oxidative stress with associated inflammation, alveolar epithelial cell apoptosis, surfactant dysfunction, pulmonary vascular abnormalities, resorption atelectasis, and impairment of innate immunity predisposing to secondary bacterial infections. While oxygen is a life-saving treatment, alveolar hyperoxia may exacerbate pre-existing lung injury.
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a novel member of the enveloped ribonucleic acid (RNA) beta coronavirus family, is the infectious agent causing the coronavirus disease 2019 (COVID-19) pandemic that has resulted in significant health, social, and economic burdens worldwide [1]. Severe COVID-19 disease is associated with male gender predominance, older age, metabolic syndrome, and obesity [2]. Moreover, some patients develop a multi-system disease process involving major organs with acute myocardial injury, acute kidney injury, haematological abnormalities, and intracerebral complications with prothrombotic tendencies [3]. A minority of hospitalised patients with severe COVID-19 pneumonia develop acute hypoxaemic respiratory failure (AHRF) necessitating intensive care admission and the initiation of mechanical ventilation to support adequate arterial oxygenation [4]. As the management of these critically ill patients continues to evolve, the pathophysiology of severe COVID-19 lung injury remains an intriguing phenomenon [5]. Although COVID-19 is defined as a single disease entity with a single causative agent, diverse clinical phenotypes may warrant individualised treatment approaches. These phenotypes are likely to reflect the complex host–virus interaction associated with SARS-CoV-2 infection, particularly the degree of host immunothrombotic response during and after the viral illness [6]. There is often a lag of 7–12 days between infection and the development of AHRF with SARS-CoV-2 infection, and antiviral strategies at this stage appear to offer no survival advantage [7][8]. Some patients go on to develop sustained hypoxaemic respiratory failure with prolonged hospitalisations.
2. Ubiquitous Use of Oxygen May Be a Problem—Pre COVID-19 Studies
3. Oxygen Toxicity Mechanisms
4. Alveolar Hyperoxia Induced Surfactant Damage
5. Alveolar Hyperoxia and the Expression of ACE2 Receptors
6. Alveolar Hyperoxia Induced Pulmonary Vascular Changes
7. Hyperoxia Induced Immune Dysfunction and Secondary Bacterial Infections
References
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 21 September 2023).
- Wingert, A.; Pillay, J.; Gates, M.; Guitard, S.; Rahman, S.; Beck, A.; Vandermeer, B.; Hartling, L. Risk factors for severity of COVID-19: A rapid review to inform vaccine prioritisation in Canada. BMJ Open 2021, 11, e044684.
- Roberts, C.M.; Levi, M.; McKee, M.; Schilling, R.; Lim, W.S.; Grocott, M.P.W. COVID-19: A complex multisystem disorder. Br. J. Anaesth. 2020, 125, 238–242.
- Wunsch, H. Mechanical Ventilation in COVID-19: Interpreting the Current Epidemiology. Am. J. Respir. Crit. Care Med. 2020, 202, 1–4.
- Osuchowski, M.F.; Winkler, M.S.; Skirecki, T.; Cajander, S.; Shankar-Hari, M.; Lachmann, G.; Monneret, G.; Venet, F.; Bauer, M.; Brunkhorst, F.M.; et al. The COVID-19 puzzle: Deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir. Med. 2021, 9, 622–642.
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated with Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189.
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062.
- Li, L.; Li, R.; Wu, Z.; Yang, X.; Zhao, M.; Liu, J.; Chen, D. Therapeutic strategies for critically ill patients with COVID-19. Ann. Intensive Care 2020, 10, 45.
- Clark, J.M.; Lambertsen, C.J. Pulmonary oxygen toxicity: A review. Pharmacol. Rev. 1971, 23, 37–133.
- Smith, J.L. The pathological effects due to increase of oxygen tension in the air breathed. J. Physiol. 1899, 24, 19–35.
- Kallet, R.H.; Matthay, M.A. Hyperoxic acute lung injury. Respir. Care 2013, 58, 123–141.
- Whitehead, G.S.; Burch, L.H.; Berman, K.G.; Piantadosi, C.A.; Schwartz, D.A. Genetic basis of murine responses to hyperoxia-induced lung injury. Immunogenetics 2006, 58, 793–804.
- Bhandari, V.; Elias, J.A. Cytokines in tolerance to hyperoxia-induced injury in the developing and adult lung. Free. Radic. Biol. Med. 2006, 41, 4–18.
- Coalson, J.J.; King, R.J.; Winter, V.T.; Prihoda, T.J.; Anzueto, A.R.; Peters, J.I.; Johanson, W.G., Jr. O2− and pneumonia-induced lung injury. I. Pathological and morphometric studies. J. Appl. Physiol. 1989, 67, 346–356.
- Fracica, P.J.; Caminiti, S.P.; Piantadosi, C.A.; Duhaylongsod, F.G.; Crapo, J.D.; Young, S.L. Natural surfactant and hyperoxic lung injury in primates. II. Morphometric analyses. J. Appl. Physiol. 1994, 76, 1002–1010.
- Comroe, J.H.; Dripps, R.D.; Dumke, P.R.; Deming, M. Oxygen toxicity: The effect of inhalation of high concentrations of oxygen for twenty-four hours on normal men at sea level and at a simulated altitude of 18,000 feet. J. Am. Med. Assoc. 1945, 128, 710–717.
- Caldwell, P.R.; Lee, W.L., Jr.; Schildkraut, H.S.; Archibald, E.R. Changes in lung volume, diffusing capacity, and blood gases in men breathing oxygen. J. Appl. Physiol. 1966, 21, 1477–1483.
- Sackner, M.A.; Landa, J.; Hirsch, J.; Zapata, A. Pulmonary effects of oxygen breathing. A 6-hour study in normal men. Ann. Intern. Med. 1975, 82, 40–43.
- Davis, W.B.; Rennard, S.I.; Bitterman, P.B.; Crystal, R.G. Pulmonary oxygen toxicity. Early reversible changes in human alveolar structures induced by hyperoxia. N. Engl. J. Med. 1983, 309, 878–883.
- Tateda, K.; Deng, J.C.; Moore, T.A.; Newstead, M.W.; Paine, R., 3rd; Kobayashi, N.; Yamaguchi, K.; Standiford, T.J. Hyperoxia mediates acute lung injury and increased lethality in murine Legionella pneumonia: The role of apoptosis. J. Immunol. 2003, 170, 4209–4216.
- Chu, D.K.; Kim, L.H.; Young, P.J.; Zamiri, N.; Almenawer, S.A.; Jaeschke, R.; Szczeklik, W.; Schunemann, H.J.; Neary, J.D.; Alhazzani, W. Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): A systematic review and meta-analysis. Lancet 2018, 391, 1693–1705.
- Schjorring, O.L.; Klitgaard, T.L.; Perner, A.; Wetterslev, J.; Lange, T.; Siegemund, M.; Backlund, M.; Keus, F.; Laake, J.H.; Morgan, M.; et al. Lower or Higher Oxygenation Targets for Acute Hypoxemic Respiratory Failure. N. Engl. J. Med. 2021, 384, 1301–1311.
- Barrot, L.; Asfar, P.; Mauny, F.; Winiszewski, H.; Montini, F.; Badie, J.; Quenot, J.P.; Pili-Floury, S.; Bouhemad, B.; Louis, G.; et al. Liberal or Conservative Oxygen Therapy for Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2020, 382, 999–1008.
- Protocol and Statistical Analysis Plan for the Mega Randomised Registry Trial Research Program Comparing Conservative versus Liberal Oxygenation Targets in Adults Receiving Unplanned Invasive Mechanical Ventilation in the ICU (Mega-ROX)|CCR. 7 July 2022. Available online: https://ccr.cicm.org.au/journal-editions/2022/june/24/original-articles/article-8 (accessed on 21 September 2023).
- UK-ROX. Intensive Care Unit Randomised Trial Comparing Two Approaches to OXygen Therapy. 7 July 2022. Available online: https://www.icnarc.org/Our-Research/Studies/Uk-Rox/About (accessed on 21 September 2023).
- Mega-ROX. In ANZICS . 8 July 2022. Available online: https://www.anzics.com.au/current-active-endorsed-research/mega-rox/ (accessed on 24 September 2023).
- Rowe, L.A.; Degtyareva, N.; Doetsch, P.W. DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae. Free Radic. Biol. Med. 2008, 45, 1167–1177.
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392.
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293.
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic. Biol. Med. 2011, 51, 1000–1013.
- Muhammad, Y.; Kani, Y.A.; Iliya, S.; Muhammad, J.B.; Binji, A.; El-Fulaty Ahmad, A.; Kabir, M.B.; Umar Bindawa, K.; Ahmed, A. Deficiency of antioxidants and increased oxidative stress in COVID-19 patients: A cross-sectional comparative study in Jigawa, Northwestern Nigeria. SAGE Open Med. 2021, 9, 2050312121991246.
- Chiscano-Camon, L.; Ruiz-Rodriguez, J.C.; Ruiz-Sanmartin, A.; Roca, O.; Ferrer, R. Vitamin C levels in patients with SARS-CoV-2-associated acute respiratory distress syndrome. Crit. Care 2020, 24, 522.
- Pincemail, J.; Cavalier, E.; Charlier, C.; Cheramy-Bien, J.P.; Brevers, E.; Courtois, A.; Fadeur, M.; Meziane, S.; Goff, C.L.; Misset, B.; et al. Oxidative Stress Status in COVID-19 Patients Hospitalized in Intensive Care Unit for Severe Pneumonia. A Pilot Study. Antioxidants 2021, 10, 257.
- Sena, C.M.; Leandro, A.; Azul, L.; Seica, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668.
- Porzionato, A.; Sfriso, M.M.; Mazzatenta, A.; Macchi, V.; De Caro, R.; Di Giulio, C. Effects of hyperoxic exposure on signal transduction pathways in the lung. Respir. Physiol. Neurobiol. 2015, 209, 106–114.
- Dias-Freitas, F.; Metelo-Coimbra, C.; Roncon-Albuquerque, R., Jr. Molecular mechanisms underlying hyperoxia acute lung injury. Respir. Med. 2016, 119, 23–28.
- Walsh, C.M.; Luhrs, K.A.; Arechiga, A.F. The “fuzzy logic” of the death-inducing signaling complex in lymphocytes. J. Clin. Immunol. 2003, 23, 333–353.
- Cho, H.Y.; Jedlicka, A.E.; Reddy, S.P.; Kensler, T.W.; Yamamoto, M.; Zhang, L.Y.; Kleeberger, S.R. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2002, 26, 175–182.
- Cho, H.Y.; Kleeberger, S.R. Nrf2 protects against airway disorders. Toxicol. Appl. Pharmacol. 2010, 244, 43–56.
- Dushianthan, A.; Goss, V.; Cusack, R.; Grocott, M.P.; Postle, A.D. Phospholipid composition and kinetics in different endobronchial fractions from healthy volunteers. BMC Pulm. Med. 2014, 14, 10.
- Dushianthan, A.; Goss, V.; Cusack, R.; Grocott, M.P.; Postle, A.D. Altered molecular specificity of surfactant phosphatidycholine synthesis in patients with acute respiratory distress syndrome. Respir. Res. 2014, 15, 128.
- Andersson, S.; Kheiter, A.; Merritt, T.A. Oxidative inactivation of surfactants. Lung 1999, 177, 179–189.
- Rodriguez-Capote, K.; Manzanares, D.; Haines, T.; Possmayer, F. Reactive oxygen species inactivation of surfactant involves structural and functional alterations to surfactant proteins SP-B and SP-C. Biophys. J. 2006, 90, 2808–2821.
- Tolle, A.; Kolleck, I.; Schlame, M.; Wauer, R.; Stevens, P.A.; Rustow, B. Effect of hyperoxia on the composition of the alveolar surfactant and the turnover of surfactant phospholipids, cholesterol, plasmalogens and vitamin E. Biochim. Biophys. Acta 1997, 1346, 198–204.
- Pace, P.W.; Yao, L.J.; Wilson, J.X.; Possmayer, F.; Veldhuizen, R.A.; Lewis, J.F. The effects of hyperoxia exposure on lung function and pulmonary surfactant in a rat model of acute lung injury. Exp. Lung Res. 2009, 35, 380–398.
- Putman, E.; van Golde, L.M.; Haagsman, H.P. Toxic oxidant species and their impact on the pulmonary surfactant system. Lung 1997, 175, 75–103.
- Calkovska, A.; Kolomaznik, M.; Calkovsky, V. Alveolar type II cells and pulmonary surfactant in COVID-19 era. Physiol. Res. 2021, 70 (Suppl. S2), S195–S208.
- Kennedy, K.A.; Snyder, J.M.; Stenzel, W.; Saito, K.; Warshaw, J.B. Vitamin E alters alveolar type II cell phospholipid synthesis in oxygen and air. Exp. Lung Res. 1990, 16, 607–615.
- Polak, M.J.; Knight, M.E.; Andresen, T.L.; DeSena, C. Effects of hyperoxia and beta-adrenergic stimulation on pulmonary surfactant in neonatal rabbits. Exp. Lung Res. 1992, 18, 373–384.
- Jin, Y.; Peng, L.Q.; Zhao, A.L. Hyperoxia induces the apoptosis of alveolar epithelial cells and changes of pulmonary surfactant proteins. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 492–497.
- Smallwood, C.D.; Boloori-Zadeh, P.; Silva, M.R.; Gouldstone, A. High Oxygen Concentrations Adversely Affect the Performance of Pulmonary Surfactant. Respir. Care 2017, 62, 1085–1090.
- Meng, H.; Sun, Y.; Lu, J.; Fu, S.; Meng, Z.; Scott, M.; Li, Q. Exogenous surfactant may improve oxygenation but not mortality in adult patients with acute lung injury/acute respiratory distress syndrome: A meta-analysis of 9 clinical trials. J. Cardiothorac. Vasc. Anesth. 2012, 26, 849–856.
- Postle, A.D.; Clark, H.W.; Fink, J.; Madsen, J.; Koster, G.; Panchal, M.; Djukanovic, R.; Brealey, D.; Grocott, M.P.W.; Dushianthan, A. Rapid Phospholipid Turnover after Surfactant Nebulization in Severe COVID-19 Infection: A Randomized Clinical Trial. Am. J. Respir. Crit. Care Med. 2022, 205, 471–473.
- Engstrom, P.C.; Holm, B.A.; Matalon, S. Surfactant replacement attenuates the increase in alveolar permeability in hyperoxia. J. Appl. Physiol. 1989, 67, 688–693.
- Matalon, S.; Holm, B.A.; Notter, R.H. Mitigation of pulmonary hyperoxic injury by administration of exogenous surfactant. J. Appl. Physiol. 1987, 62, 756–761.
- Bezerra, F.S.; Ramos, C.O.; Castro, T.F.; Araujo, N.; de Souza, A.B.F.; Bandeira, A.C.B.; Costa, G.P.; Cartelle, C.T.; Talvani, A.; Cangussu, S.D.; et al. Exogenous surfactant prevents hyperoxia-induced lung injury in adult mice. Intensive Care Med. Exp. 2019, 7, 19.
- Dani, C.; Corsini, I.; Longini, M.; Burchielli, S.; Dichiara, G.; Cantile, C.; Buonocore, G. Natural surfactant combined with superoxide dismutase and catalase decreases oxidative lung injury in the preterm lamb. Pediatr. Pulmonol. 2014, 49, 898–904.
- Ghio, A.J.; Fracica, P.J.; Young, S.L.; Piantadosi, C.A. Synthetic surfactant scavenges oxidants and protects against hyperoxic lung injury. J. Appl. Physiol. 1994, 77, 1217–1223.
- Srivastava, S.; Garg, I.; Bansal, A.; Kumar, B. SARS-CoV-2 infection: Physiological and environmental gift factors at high altitude. Virusdisease 2020, 31, 450–452.
- Pun, M.; Turner, R.; Strapazzon, G.; Brugger, H.; Swenson, E.R. Lower Incidence of COVID-19 at High Altitude: Facts and Confounders. High Alt. Med. Biol. 2020, 21, 217–222.
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.J.; Xie, J.; Liu, Y.M.; Zhao, Y.C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality Among Patients with Hypertension Hospitalized With COVID-19. Circ. Res. 2020, 126, 1671–1681.
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621.
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L84–L96.
- Li, X.; Molina-Molina, M.; Abdul-Hafez, A.; Uhal, V.; Xaubet, A.; Uhal, B.D. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L178–L185.
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116.
- Fang, Y.; Gao, F.; Liu, Z. Angiotensin-converting enzyme 2 attenuates inflammatory response and oxidative stress in hyperoxic lung injury by regulating NF-kappaB and Nrf2 pathways. QJM 2019, 112, 914–924.
- Oarhe, C.I.; Dang, V.; Dang, M.; Nguyen, H.; Gopallawa, I.; Gewolb, I.H.; Uhal, B.D. Hyperoxia downregulates angiotensin-converting enzyme-2 in human fetal lung fibroblasts. Pediatr. Res. 2015, 77, 656–662.
- Writing Committee for the REMAP-CAP Investigators; Lawler, P.R.; Derde, L.P.G.; van de Veerdonk, F.L.; McVerry, B.J.; Huang, D.T.; Berry, L.R.; Lorenzi, E.; van Kimmenade, R.; Gommans, F.; et al. Effect of Angiotensin-Converting Enzyme Inhibitor and Angiotensin Receptor Blocker Initiation on Organ Support-Free Days in Patients Hospitalized With COVID-19: A Randomized Clinical Trial. JAMA 2023, 329, 1183–1196.
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128.
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418.
- Six, S.; Jaffal, K.; Ledoux, G.; Jaillette, E.; Wallet, F.; Nseir, S. Hyperoxemia as a risk factor for ventilator-associated pneumonia. Crit. Care 2016, 20, 195.
- Maes, M.; Higginson, E.; Pereira-Dias, J.; Curran, M.D.; Parmar, S.; Khokhar, F.; Cuchet-Lourenco, D.; Lux, J.; Sharma-Hajela, S.; Ravenhill, B.; et al. Ventilator-associated pneumonia in critically ill patients with COVID-19. Crit. Care 2021, 25, 25.
- Rawson, T.M.; Moore, L.S.P.; Zhu, N.; Ranganathan, N.; Skolimowska, K.; Gilchrist, M.; Satta, G.; Cooke, G.; Holmes, A. Bacterial and Fungal Coinfection in Individuals with Coronavirus: A Rapid Review to Support COVID-19 Antimicrobial Prescribing. Clin. Infect. Dis. 2020, 71, 2459–2468.
- O’Reilly, P.J.; Hickman-Davis, J.M.; Davis, I.C.; Matalon, S. Hyperoxia impairs antibacterial function of macrophages through effects on actin. Am. J. Respir. Cell Mol. Biol. 2003, 28, 443–450.
- Gauthier, A.G.; Wu, J.; Lin, M.; Sitapara, R.; Kulkarni, A.; Thakur, G.A.; Schmidt, E.E.; Perron, J.C.; Ashby, C.R., Jr.; Mantell, L.L. The Positive Allosteric Modulation of alpha7-Nicotinic Cholinergic Receptors by GAT107 Increases Bacterial Lung Clearance in Hyperoxic Mice by Decreasing Oxidative Stress in Macrophages. Antioxidants 2021, 10, 135.
- Baleeiro, C.E.; Wilcoxen, S.E.; Morris, S.B.; Standiford, T.J.; Paine, R., 3rd. Sublethal hyperoxia impairs pulmonary innate immunity. J. Immunol. 2003, 171, 955–963.
- Ashley, S.L.; Sjoding, M.W.; Popova, A.P.; Cui, T.X.; Hoostal, M.J.; Schmidt, T.M.; Branton, W.R.; Dieterle, M.G.; Falkowski, N.R.; Baker, J.M.; et al. Lung and gut microbiota are altered by hyperoxia and contribute to oxygen-induced lung injury in mice. Sci. Transl. Med. 2020, 12, eaau9959.
- Xing, Z.; Li, Y.; Liu, G.; He, Y.; Tao, Y.; Chen, M. Hyperoxia provokes gut dysbiosis in rats. Crit. Care 2020, 24, 517.


