 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohammad Islam | -- | 3339 | 2023-11-16 10:34:27 | | | |
| 2 | Lindsay Dong | Meta information modification | 3339 | 2023-11-20 02:21:13 | | |
Video Upload Options
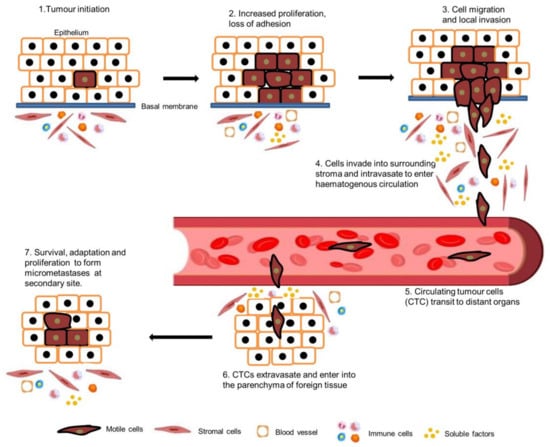
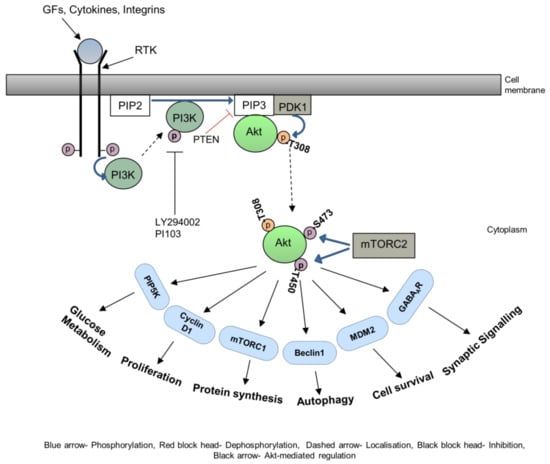
Metastasis is a critical step in the process of carcinogenesis and a vast majority of cancer-related mortalities result from metastatic disease that is resistant to current therapies. Cell migration and invasion are the first steps of the metastasis process, which mainly occurs by two important biological mechanisms, i.e., cytoskeletal remodelling and epithelial to mesenchymal transition (EMT). Akt (also known as protein kinase B) is a central signalling molecule of the PI3K-Akt signalling pathway. Aberrant activation of this pathway has been identified in a wide range of cancers. Several studies have revealed that Akt actively engages with the migratory process in motile cells, including metastatic cancer cells. The downstream signalling mechanism of Akt in cell migration depends upon the tumour type, sites, and intracellular localisation of activated Akt.
1. Introduction
2. Akt in Cytoskeletal Rearrangements
3. Akt in EMT
4. Akt in HNSCC Metastasis
5. Conclusions
References
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576.
- Palmer, T.D.; Ashby, W.J.; Lewis, J.D.; Zijlstra, A. Targeting tumor cell motility to prevent metastasis. Adv. Drug Deliv. Rev. 2011, 63, 568–581.
- Leber, M.F.; Efferth, T. Molecular principles of cancer invasion and metastasis (review). Int. J. Oncol. 2009, 34, 881–895.
- Robert, J. Biology of cancer metastasis. Bull. Cancer 2013, 100, 333–342.
- Woodhouse, E.C.; Chuaqui, R.F.; Liotta, L.A. General mechanisms of metastasis. Cancer 1997, 80, 1529–1537.
- Luo, W. Nasopharyngeal carcinoma ecology theory: Cancer as multidimensional spatiotemporal “unity of ecology and evolution” pathological ecosystem. Theranostics 2023, 13, 1607–1631.
- Friedl, P.; Brocker, E.B. The biology of cell locomotion within three-dimensional extracellular matrix. Cell. Mol. Life Sci. 2000, 57, 41–64.
- Lauffenburger, D.A.; Horwitz, A.F. Cell migration: A physically integrated molecular process. Cell 1996, 84, 359–369.
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell Migration: Integrating Signals from Front to Back. Science 2003, 302, 1704–1709.
- Mitchison, T.J.; Cramer, L.P. Actin-based cell motility and cell locomotion. Cell 1996, 84, 371–379.
- Ahmed, H.; Ghoshal, A.; Jones, S.; Ellis, I.; Islam, M. Head and Neck Cancer Metastasis and the Effect of the Local Soluble Factors, from the Microenvironment, on Signalling Pathways: Is It All about the Akt? Cancers 2020, 12, 2093.
- Ellis, I.R.; Islam, M.R.; Aljorani, L.; Jones, S.J. Fibronectin: The N-terminal region and its role in cell migration- implications for disease and healing. In Fibronectin: Current Concepts in Structure, Function and Pathology; Beattie, J., Ed.; Protein Biochemistry, Synthesis, Structure and Cellular Functions; Nova Science Publishers: New York, NY, USA, 2012; pp. 35–69.
- Friedl, P. Prespecification and plasticity: Shifting mechanisms of cell migration. Curr. Opin. Cell Biol. 2004, 16, 14–23.
- Inaki, M.; Vishnu, S.; Cliffe, A.; Rorth, P. Effective guidance of collective migration based on differences in cell states. Proc. Natl. Acad. Sci. USA 2012, 109, 2027–2032.
- Rorth, P. Whence directionality: Guidance mechanisms in solitary and collective cell migration. Dev. Cell 2011, 20, 9–18.
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374.
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783.
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274.
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927.
- Alessi, D.R.; Cohen, P. Mechanism of activation and function of protein kinase B. Curr. Opin. Genet. Dev. 1998, 8, 55–62.
- Bozulic, L.; Hemmings, B.A. PIKKing on PKB: Regulation of PKB activity by phosphorylation. Curr. Opin. Cell Biol. 2009, 21, 256–261.
- Feng, J.; Park, J.; Cron, P.; Hess, D.; Hemmings, B.A. Identification of a PKB/Akt Hydrophobic Motif Ser-473 Kinase as DNA-dependent Protein Kinase. J. Biol. Chem. 2004, 279, 41189–41196.
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101.
- Bellacosa, A.; Chan, T.O.; Ahmed, N.N.; Datta, K.; Malstrom, S.; Stokoe, D.; McCormick, F.; Feng, J.; Tsichlis, P. Akt activation by growth factors is a multiple-step process: The role of the PH domain. Oncogene 1998, 17, 313–325.
- Hart, J.R.; Vogt, P.K. Phosphorylation of AKT: A mutational analysis. Oncotarget 2011, 2, 467–476.
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.-L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. Available online: http://www.nature.com/emboj/journal/v27/n14/suppinfo/emboj2008119a_S1.html (accessed on 5 September 2023).
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644.
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959.
- De Marco, C.; Rinaldo, N.; Bruni, P.; Malzoni, C.; Zullo, F.; Fabiani, F.; Losito, S.; Scrima, M.; Marino, F.Z.; Franco, R.; et al. Multiple genetic alterations within the PI3K pathway are responsible for AKT activation in patients with ovarian carcinoma. PLoS ONE 2013, 8, e55362.
- Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Stemke-Hale, K.; Sahin, A.; Liu, S.; Barrera, J.A.; Burgues, O.; Lluch, A.M.; Chen, H.; Hortobagyi, G.N. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol. Cancer Ther. 2011, 10, 1093–1101.
- Wu, R.; Baker, S.J.; Hu, T.C.; Norman, K.M.; Fearon, E.R.; Cho, K.R. Type I to Type II Ovarian Carcinoma Progression: Mutant Trp53 or Pik3ca Confers a More Aggressive Tumor Phenotype in a Mouse Model of Ovarian Cancer. Am. J. Pathol. 2013, 182, 1391–1399.
- Xue, G.; Hemmings, B.A. PKB/Akt-Dependent Regulation of Cell Motility. J. Natl. Cancer Inst. 2013, 105, 393–404.
- Yoeli-Lerner, M.; Toker, A. Akt/PKB Signaling in Cancer: A Function in Cell Motility and Invasion. Cell Cycle 2006, 5, 603–605.
- Frixione, E. Recurring views on the structure and function of the cytoskeleton: A 300-year epic. Cell Motil. Cytoskelet. 2000, 46, 73–94.
- Bonello, T.; Coombes, J.; Schevzov, G.; Gunning, P.; Stehn, J. Therapeutic Targeting of the Actin Cytoskeleton in Cancer. In Cytoskeleton and Human Disease; Kavallaris, M., Ed.; Humana Press: New York, NY, USA, 2012; pp. 181–200.
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465.
- Bugyi, B.; Carlier, M.F. Control of actin filament treadmilling in cell motility. Annu. Rev. Biophys. 2010, 39, 449–470.
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31.
- Morales-Ruiz, M.; Fulton, D.; Sowa, G.; Languino, L.R.; Fujio, Y.; Walsh, K.; Sessa, W.C. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ. Res. 2000, 86, 892–896.
- Qian, Y.; Corum, L.; Meng, Q.; Blenis, J.; Zheng, J.Z.; Shi, X.; Flynn, D.C.; Jiang, B.H. PI3K induced actin filament remodeling through Akt and p70S6K1: Implication of essential role in cell migration. Am. J. Physiol. Cell Physiol. 2004, 286, C153–C163.
- Chhabra, E.S.; Higgs, H.N. The many faces of actin: Matching assembly factors with cellular structures. Nat. Cell Biol. 2007, 9, 1110–1121.
- Enomoto, A.; Murakami, H.; Asai, N.; Morone, N.; Watanabe, T.; Kawai, K.; Murakumo, Y.; Usukura, J.; Kaibuchi, K.; Takahashi, M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev. Cell 2005, 9, 389–402.
- Feng, Y.; Walsh, C.A. The many faces of filamin: A versatile molecular scaffold for cell motility and signalling. Nat. Cell Biol. 2004, 6, 1034–1038.
- Ravid, D.; Chuderland, D.; Landsman, L.; Lavie, Y.; Reich, R.; Liscovitch, M. Filamin A is a novel caveolin-1-dependent target in IGF-I-stimulated cancer cell migration. Exp. Cell Res. 2008, 314, 2762–2773.
- Stossel, T.P.; Condeelis, J.; Cooley, L.; Hartwig, J.H.; Noegel, A.; Schleicher, M.; Shapiro, S.S. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2001, 2, 138–145.
- Ravid, D.; Maor, S.; Werner, H.; Liscovitch, M. Caveolin-1 inhibits cell detachment-induced p53 activation and anoikis by upregulation of insulin-like growth factor-I receptors and signaling. Oncogene 2005, 24, 1338–1347.
- Nallapalli, R.K.; Ibrahim, M.X.; Zhou, A.X.; Bandaru, S.; Sunkara, S.N.; Redfors, B.; Pazooki, D.; Zhang, Y.; Boren, J.; Cao, Y.; et al. Targeting filamin A reduces K-RAS-induced lung adenocarcinomas and endothelial response to tumor growth in mice. Mol. Cancer 2012, 11, 50.
- Meima, M.E.; Webb, B.A.; Witkowska, H.E.; Barber, D.L. The sodium-hydrogen exchanger NHE1 is an Akt substrate necessary for actin filament reorganization by growth factors. J. Biol. Chem. 2009, 284, 26666–26675.
- Chang, L.; Goldman, R.D. Intermediate filaments mediate cytoskeletal crosstalk. Nat. Rev. Mol. Cell Biol. 2004, 5, 601–613.
- Helfand, B.T.; Chang, L.; Goldman, R.D. Intermediate filaments are dynamic and motile elements of cellular architecture. J. Cell Sci. 2004, 117, 133–141.
- Zhu, Q.S.; Rosenblatt, K.; Huang, K.L.; Lahat, G.; Brobey, R.; Bolshakov, S.; Nguyen, T.; Ding, Z.; Belousov, R.; Bill, K.; et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 2011, 30, 457–470.
- Lahat, G.; Zhu, Q.S.; Huang, K.L.; Wang, S.; Bolshakov, S.; Liu, J.; Torres, K.; Langley, R.R.; Lazar, A.J.; Hung, M.C.; et al. Vimentin is a novel anti-cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS ONE 2010, 5, e10105.
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol. Life Sci. 2011, 68, 3033–3046.
- Gao, D.; Inuzuka, H.; Tseng, A.; Chin, R.Y.; Toker, A.; Wei, W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat. Cell Biol. 2009, 11, 397–408.
- Lin, H.K.; Wang, G.; Chen, Z.; Teruya-Feldstein, J.; Liu, Y.; Chan, C.H.; Yang, W.L.; Erdjument-Bromage, H.; Nakayama, K.I.; Nimer, S.; et al. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat. Cell Biol. 2009, 11, 420–432.
- Dimmeler, S.; Dernbach, E.; Zeiher, A.M. Phosphorylation of the endothelial nitric oxide synthase at ser-1177 is required for VEGF-induced endothelial cell migration. FEBS Lett. 2000, 477, 258–262.
- Ridnour, L.A.; Barasch, K.M.; Windhausen, A.N.; Dorsey, T.H.; Lizardo, M.M.; Yfantis, H.G.; Lee, D.H.; Switzer, C.H.; Cheng, R.Y.; Heinecke, J.L.; et al. Nitric oxide synthase and breast cancer: Role of TIMP-1 in NO-mediated Akt activation. PLoS ONE 2012, 7, e44081.
- Xu, W.; Liu, L.Z.; Loizidou, M.; Ahmed, M.; Charles, I.G. The role of nitric oxide in cancer. Cell Res. 2002, 12, 311–320.
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407.
- Lee, M.J.; Thangada, S.; Paik, J.H.; Sapkota, G.P.; Ancellin, N.; Chae, S.S.; Wu, M.; Morales-Ruiz, M.; Sessa, W.C.; Alessi, D.R.; et al. Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Mol. Cell 2001, 8, 693–704.
- Ozaki, H.; Hla, T.; Lee, M.J. Sphingosine-1-phosphate signaling in endothelial activation. J. Atheroscler. Thromb. 2003, 10, 125–131.
- Li, J.; Ballif, B.A.; Powelka, A.M.; Dai, J.; Gygi, S.P.; Hsu, V.W. Phosphorylation of ACAP1 by Akt regulates the stimulation-dependent recycling of integrin beta1 to control cell migration. Dev. Cell 2005, 9, 663–673.
- Rowland, A.F.; Larance, M.; Hughes, W.E.; James, D.E. Identification of RhoGAP22 as an Akt-dependent regulator of cell motility in response to insulin. Mol. Cell Biol. 2011, 31, 4789–4800.
- Berven, L.A.; Willard, F.S.; Crouch, M.F. Role of the p70(S6K) pathway in regulating the actin cytoskeleton and cell migration. Exp. Cell Res. 2004, 296, 183–195.
- Sakakibara, K.; Liu, B.; Hollenbeck, S.; Kent, K.C. Rapamycin inhibits fibronectin-induced migration of the human arterial smooth muscle line (E47) through the mammalian target of rapamycin. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2861–H2868.
- Nieto, M.A. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376.
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829.
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3’ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454.
- Zheng, H.; Kang, Y. Multilayer control of the EMT master regulators. Oncogene 2013, 33, 1755–1763.
- Pal, A.; Barrett, T.F.; Paolini, R.; Parikh, A.; Puram, S.V. Partial EMT in head and neck cancer biology: A spectrum instead of a switch. Oncogene 2021, 40, 5049–5065.
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264.
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226.
- Thompson, E.W.; Williams, E.D. EMT and MET in carcinoma--clinical observations, regulatory pathways and new models. Clin. Exp. Metastasis 2008, 25, 591–592.
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of AKT kinases in cancer: Implications for therapeutic targeting. Adv. Cancer Res. 2005, 94, 29–86.
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204.
- Ringel, M.D.; Hayre, N.; Saito, J.; Saunier, B.; Schuppert, F.; Burch, H.; Bernet, V.; Burman, K.D.; Kohn, L.D.; Saji, M. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001, 61, 6105–6111.
- Testa, J.R.; Bellacosa, A. AKT plays a central role in tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 10983–10985.
- Wu, H.T.; Ko, S.Y.; Fong, J.H.; Chang, K.W.; Liu, T.Y.; Kao, S.Y. Expression of phosphorylated Akt in oral carcinogenesis and its induction by nicotine and alkaline stimulation. J. Oral Pathol. Med. 2009, 38, 206–213.
- Bellacosa, A.; de Feo, D.; Godwin, A.K.; Bell, D.W.; Cheng, J.Q.; Altomare, D.A.; Wan, M.; Dubeau, L.; Scambia, G.; Masciullo, V.; et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int. J. Cancer 1995, 64, 280–285.
- Grille, S.J.; Bellacosa, A.; Upson, J.; Klein-Szanto, A.J.; Van Roy, F.; Lee-Kwon, W.; Donowitz, M.; Larue, L. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 2003, 63, 2172–2178.
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428.
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890.
- Bellacosa, A.; Larue, L. PI3K/AKT Pathway and the Epithelial–Mesenchymal Transition. In Cancer Genome and Tumor Microenvironment; Thomas-Tikhonenko, A., Ed.; Springer Science+Business Media: New York, NY, USA, 2010; pp. 11–31.
- Katoh, M.; Katoh, M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol. Ther. 2006, 5, 1059–1064.
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940.
- Evdokimova, V.; Tognon, C.; Ng, T.; Ruzanov, P.; Melnyk, N.; Fink, D.; Sorokin, A.; Ovchinnikov, L.P.; Davicioni, E.; Triche, T.J.; et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell 2009, 15, 402–415.
- Villagrasa, P.; Diaz, V.M.; Vinas-Castells, R.; Peiro, S.; Del Valle-Perez, B.; Dave, N.; Rodriguez-Asiain, A.; Casal, J.I.; Lizcano, J.M.; Dunach, M.; et al. Akt2 interacts with Snail1 in the E-cadherin promoter. Oncogene 2012, 31, 4022–4033.
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987.
- Hong, K.O.; Kim, J.H.; Hong, J.S.; Yoon, H.J.; Lee, J.I.; Hong, S.P.; Hong, S.D. Inhibition of Akt activity induces the mesenchymal-to-epithelial reverting transition with restoring E-cadherin expression in KB and KOSCC-25B oral squamous cell carcinoma cells. J. Exp. Clin. Cancer Res. CR 2009, 28, 28.
- Xue, G.; Restuccia, D.F.; Lan, Q.; Hynx, D.; Dirnhofer, S.; Hess, D.; Ruegg, C.; Hemmings, B.A. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-beta signaling axes. Cancer Discov. 2012, 2, 248–259.
- Yao, K.; Ye, P.P.; Tan, J.; Tang, X.J.; Shen Tu, X.C. Involvement of PI3K/Akt pathway in TGF-beta2-mediated epithelial mesenchymal transition in human lens epithelial cells. Ophthalmic Res. 2008, 40, 69–76.
- Yokoyama, K.; Kimoto, K.; Itoh, Y.; Nakatsuka, K.; Matsuo, N.; Yoshioka, H.; Kubota, T. The PI3K/Akt pathway mediates the expression of type I collagen induced by TGF-beta2 in human retinal pigment epithelial cells. Graefe’s Arch. Clin. Exp. Ophthalmol. 2012, 250, 15–23.
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992.
- Nacerddine, K.; Beaudry, J.B.; Ginjala, V.; Westerman, B.; Mattiroli, F.; Song, J.Y.; van der Poel, H.; Ponz, O.B.; Pritchard, C.; Cornelissen-Steijger, P.; et al. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. J. Clin. Investig. 2012, 122, 1920–1932.
- Guo, B.H.; Feng, Y.; Zhang, R.; Xu, L.H.; Li, M.Z.; Kung, H.F.; Song, L.B.; Zeng, M.S. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol. Cancer 2011, 10, 10.
- Song, L.B.; Li, J.; Liao, W.T.; Feng, Y.; Yu, C.P.; Hu, L.J.; Kong, Q.L.; Xu, L.H.; Zhang, X.; Liu, W.L.; et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J. Clin. Investig. 2009, 119, 3626–3636.
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 2000, 92, 709–720.
- Neville, B.W.; Day, T.A. Oral cancer and precancerous lesions. CA Cancer J. Clin. 2002, 52, 195–215.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Islam, M.R.; Ellis, I.R.; Macluskey, M.; Cochrane, L.; Jones, S.J. Activation of Akt at T308 and S473 in alcohol, tobacco and HPV-induced HNSCC: Is there evidence to support a prognostic or diagnostic role? Exp. Hematol. Oncol. 2014, 3, 25.
- Islam, M.R.; Jones, S.J.; Macluskey, M.; Ellis, I.R. Is there a pAkt between VEGF and oral cancer cell migration? Cell. Signal. 2014, 26, 1294–1302.
- Amornphimoltham, P.; Sriuranpong, V.; Patel, V.; Benavides, F.; Conti, C.J.; Sauk, J.; Sausville, E.A.; Molinolo, A.A.; Gutkind, J.S. Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: A potential target for UCN-01. Clin. Cancer Res. 2004, 10, 4029–4037.
- Amornphimoltham, P.; Patel, V.; Molinolo, A.; Gutkind, J.S. Head and Neck Cancer and PI3K/Akt/mTOR Signaling Network: Novel Molecular Targeted Therapy. In Signaling Pathways in Squamous Cancer; Glick, A.B., Van Waes, C., Eds.; Springer Science+Business Media, LLC: New York, NY, USA, 2011; pp. 407–430.
- Marquard, F.E.; Jücker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2020, 172, 113729.
- Massarelli, E.; Liu, D.D.; Lee, J.J.; El-Naggar, A.K.; Lo Muzio, L.; Staibano, S.; De Placido, S.; Myers, J.N.; Papadimitrakopoulou, V.A. Akt activation correlates with adverse outcome in tongue cancer. Cancer 2005, 104, 2430–2436.
- Yu, Z.; Weinberger, P.M.; Sasaki, C.; Egleston, B.L.; Speier, W.F.t.; Haffty, B.; Kowalski, D.; Camp, R.; Rimm, D.; Vairaktaris, E.; et al. Phosphorylation of Akt (Ser473) predicts poor clinical outcome in oropharyngeal squamous cell cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 553–558.
- Pontes, H.A.; de Aquino Xavier, F.C.; da Silva, T.S.; Fonseca, F.P.; Paiva, H.B.; Pontes, F.S.; dos Santos Pinto, D., Jr. Metallothionein and p-Akt proteins in oral dysplasia and in oral squamous cell carcinoma: An immunohistochemical study. J. Oral Pathol. Med. 2009, 38, 644–650.
- Miyazawa, J.; Mitoro, A.; Kawashiri, S.; Chada, K.K.; Imai, K. Expression of Mesenchyme-Specific Gene HMGA2 in Squamous Cell Carcinomas of the Oral Cavity. Cancer Res. 2004, 64, 2024–2029.
- Maeda, G.; Chiba, T.; Okazaki, M.; Satoh, T.; Taya, Y.; Aoba, T.; Kato, K.; Kawashiri, S.; Imai, K. Expression of SIP1 in oral squamous cell carcinomas: Implications for E-cadherin expression and tumor progression. Int. J. Oncol. 2005, 27, 1535–1541.
- Yokoyama, K.; Kamata, N.; Hayashi, E.; Hoteiya, T.; Ueda, N.; Fujimoto, R.; Nagayama, M. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol. 2001, 37, 65–71.
- Taki, M.; Kamata, N.; Yokoyama, K.; Fujimoto, R.; Tsutsumi, S.; Nagayama, M. Down-regulation of Wnt-4 and up-regulation of Wnt-5a expression by epithelial-mesenchymal transition in human squamous carcinoma cells. Cancer Sci. 2003, 94, 593–597.

