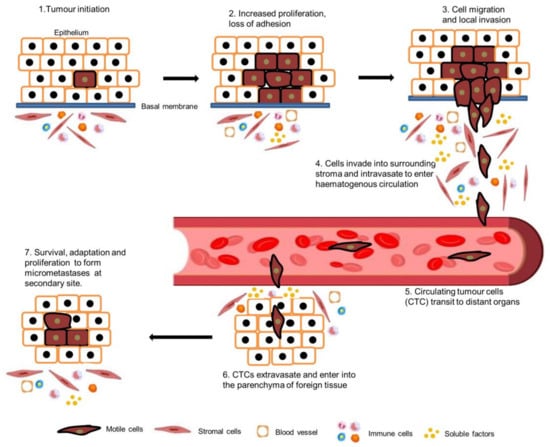
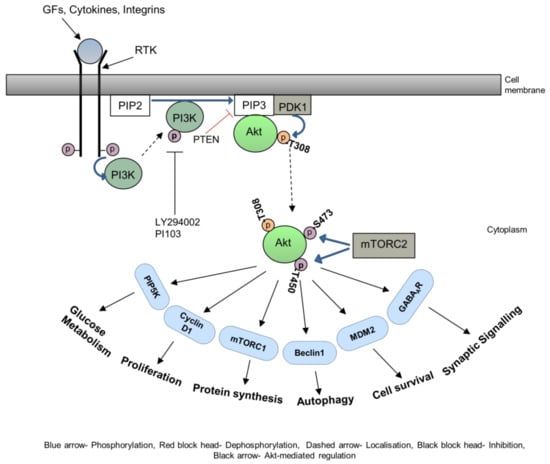
The cytoskeleton is the supporting structure of cells which is composed of a filamentous network of micro filaments such as actin and myosin, intermediate filaments such as vimentin and keratin, and microtubules such as tubulin [
37]. The main purpose of the cytoskeleton is to maintain cellular structure, intracellular transport, and supporting cell division. Cytoskeletal rearrangements occur in various physiological and pathological events such as cell movement, wound healing, and cancer metastasis [
38]. Cellular motility either in physiological events or in pathological conditions is driven by cytoskeletal remodelling, initiated by various signalling pathways. The synergistic effect of all the three basic elements—filamentous actin, microtubules, and the intermediate filament vimentin—is the potential basis for a cell to migrate [
35]. Wide-ranging studies have focused on how the stabilisation of intracellular filaments and dynamic polymerisation control cell migration [
14,
39].
Growth of the vascular network is essential for the spread of cancer cells. Angiogenesis is the process whereby new vessels are formed and involved in the supply of nutrients, oxygen, and immune cells and also the removal of waste products [
40]. Angiogenic factors play a huge role in neoplastic vascularisation, thus increasingly gaining attention. Vascular endothelial cell migration is a vital step for angiogenesis. Vascular endothelial growth factor (VEGF) activates Akt and stimulates the migration of endothelial cells by increasing actin polymerisation. Abrogated Akt activity by expression of a kinase-dead mutant inhibits actin bundle formation and blocks cell migration. This effect is enhanced when myristylated Akt is expressed [
41], demonstrating that Akt is a critical mediator of VEGF-induced endothelial cell migration through actin reorganisation.
The actin-rich structure of highly motile cells like invadopodia, filopodia, and pseudopodia needs to be stabilised to function properly. Actin-associated proteins are responsible for stabilising this actin structure by blocking the degradation of newly formed actin filaments [
54]. ALE (the Akt phosphorylation enhancer), also termed the ‘girder’ of actin filaments (Girdin), is one of the best examples of this type of protein. APE/Girdin provides the integrity of the actin meshwork (actin filament) at the leading edge of migrating cells. Reduction in APE/Girdin destabilises the actin bundles, triggering the ablation of stress fibres and actin structure. This results in the loss of directional migratory ability and establishes the vital activity of APE/Girdin in the regulation of cell migration. Enomoto et al. proved that APE/Girdin is phosphorylated by Akt on Serine 1416 (S1416) [
55].
An actin-associated structural (cross-linker) protein, filamin A, is phosphorylated by Akt on residue S2152 [
62,
63,
64]. In turn, phosphorylated filamin A mediates caveolin-1-induced cancer cell migration through the IGF signalling pathway [
65,
66]. Akt has been shown to phosphorylate NHE1 (sodium-hydrogen exchanger isoform 1), a key mediator of stress fibre disassembly on S648 and suggested to be critical for the growth factor-induced cytoskeletal rearrangements that favour cell migration and invasion [
67].
Extensive studies have been carried out to investigate the role of intermediate filaments in cell motility [
73,
74]. The most abundant intermediate protein that maintains normal cell and tissue integrity is called vimentin, a type three filamentous protein. It is phosphorylated by Akt1 on residue S39, stabilised, and thereby regulates cancer cell invasion in aggressive sarcoma [
75]. It has also been shown that vimentin is highly expressed in breast cancer lung metastases [
76,
77]; however, the specific mechanisms to control cell migration by some Akt substrates are still undefined. For example, S-phase kinase-associated protein 2 (skp2), a component of E3 ligase, is phosphorylated by Akt on the S72 residue, stimulates Skp-2 dependent ligase activity, and induces cell migration [
78,
79].
Akt interacts with promigratory proteins, in addition to targeting cytoskeletal proteins, thus facilitating crosstalk between associated signalling axes. The VEGFR/eNOS signalling pathway-controlled cell migration is dependent on Akt-mediated phosphorylation on S1177 [
42]. Accumulating evidence has indicated the importance of nitric oxide (NO) in pathological conditions, especially in malignant tumours [
83,
84]. Furthermore, VEGFR signalling often cooperates with the G-protein coupled receptor, sphingosine-1-phosphate receptor 1 (SIPR1, also known as endothelial differentiation gene 1, EDG-1). SIP/SIPR1 activation leads to the phosphor-activation of VEGFR which phosphorylates Src kinase, consequently activating the PI3K/Akt/eNOS axis [
85]. Akt-mediated phosphorylation of SIPR1 on T236 further enhances their activity and stimulates cortical actin assembly, angiogenesis, and chemotaxis [
86,
87]. Thus, Akt plays a vital role in regulating VEGFR and the SIP/SIPR1 signalling pathway and actively regulates cell migration. EphA2 (Ephrin receptor tyrosine kinase A2), a member of the largest tyrosine kinase family, is also phosphorylated by Akt on S897 residue.
It is now well established that membrane redistribution of integrin by various signalling pathways is a critical mediator of cellular movement. The ANK repeat and pleckstrin homology domain-containing protein 1 (ACAP 1) is a GTPase activating protein (GAP) for ADP ribosylation factor 6 (ARF6) known to participate in integrin beta recycling. ACAP1 is phosphorylated by Akt on S554, stimulates integrin recycling, and therefore promotes cell migration [
91]. Another GTPase activation protein, RhoGAP22, is shown to be phosphorylated by Akt on S16, upon stimulation by insulin or possibly PDGF, and this plays a vital role in regulating cell migration, leading to modulation of Rac1 activity [
92]. Various studies have established the role of the mammalian targets of rapamycin complex 1 (mTORC1) in the cell migration and relationship with Akt [
93,
94].
3. Akt in EMT
Epithelial cells are tightly connected to their adjacent cells via E-cadherin and with actin filaments via α- or β-catenin. Epithelial tumour cells must break these intercellular junctions before migrating as single cells and invading stromal tissues. Epithelial tumour cells undergo a process named epithelial to mesenchymal transition (EMT) to facilitate the invasion as a single cell. The EMT process can be induced either by extracellular growth factors, for example EGF, TGF-α and β, FGF, or by intracellular cues, such as oncogenic Ras [
110,
111]. Epithelial cells gain a mesenchymal phenotype by losing their polarity and cell–cell contacts during EMT. Functional loss of E-Cadherin and downregulation of epithelial cell markers such as cytokeratins and ZO-1, and the overexpression of mesenchymal or fibroblast cell markers such as N-cadherin, vimentin, and fibronectin are the main characteristics of EMT [
112,
113]. EMT is a complex biological process that plays a critical role in cancer metastasis. In head and neck cancer, EMT can be involved in the dissemination of cancer cells to distant sites. However, it is important to understand that EMT is not an all-or-nothing phenomenon; there are partial or hybrid states of EMT that can have unique implications for cancer metastasis.
Partial EMT (p-EMT) is a term used to describe a state in which cancer cells exhibit some, but not all, of the characteristics associated with a full EMT [
114]. p-EMT can enhance the invasive capacity of cancer cells. These cells may have an increased ability to break away from the primary tumour as a group of cells and infiltrate surrounding tissues, which is a crucial step in metastasis. Partially EMT-activated cancer cells might be less susceptible to apoptosis. This allows them to survive in the bloodstream and at distant site, where they might otherwise be eliminated by the body’s natural defences [
115,
116]. Cells which undergo p-EMT may also evade the immune system to some extent, making it more challenging for the body to recognize and destroy these cells. Partial EMT can also contribute to the formation of a pre-metastatic niche at distant sites.
EMT is reversible and, sometimes, cells undergo the reciprocal mesenchymal to epithelial transition (MET). During the development process, EMT plays an essential role in the development of various tissues and organs such as the heart, neural crest, and peripheral nervous and musculoskeletal systems. Only a small number of cells in adult organisms have the ability to go through the EMT process in specific physiological or pathological events such as wound healing. Nevertheless, tumour cells often gain the ability to reactivate the EMT process during metastasis, which enhances the migration and invasion capacity of cancer cells [
113,
120]. A number of studies have reported that Akt is frequently activated in human carcinomas [
121,
122,
123,
124,
125]. Akt2 has been shown to be activated in ovarian carcinoma, affecting epithelial cell morphology, tumorigenicity, cell motility, and invasiveness, which is characterised by the loss of histological features of epithelial differentiation [
126]. Evidence that Akt was shown to regulateEMT was first published in 2003, where squamous cell carcinoma cells, overexpressing an activated mutant of Akt, were shown to undergo EMT and downregulate E-cadherin [
127]. Loss of E-cadherin and relocalisation of β-catenin from the membrane to the nucleus is frequently detected in tumour cells undergoing EMT [
128,
129]. Several transcription factors have been recognized that can induce and maintain the EMT process, including Snail, Twist, and Zeb. The definitive molecular signalling mechanisms of normal regulation of these transcription factors are still uncertain; however, they are apparently deregulated in many invasive cancers [
112,
130]. Evidence suggests a strong relationship between Akt and EMT-inducing transcription factors. Snail is phosphorylated by GSK3β (glycogen synthase kinase 3 beta) in normal epithelial cells but is very unstable and hardly detectable. Expression of Snail in epithelial cells strongly induces morphological changes associated with enhanced migratory capacity [
131,
132].
Y-box binding protein-1 (YB-1), a transcription/translation regulatory protein, is reported to be activated by Akt and translocated to the nucleus. Nuclear YB-1 thus phosphorylates Snail and decreases E-cadherin expression, which in turn induces EMT in invasive breast carcinoma [
138]. Furthermore, upregulated Snail could, in turn, increase Akt activity. Snail increases the binding of Akt2 to the E-cadherin (CDH1) promoter and Akt2 interference unexpectedly inhibits Snail repression of the CDH1 gene [
139]. Akt2 could also be activated by another EMT-inducer, Twist, in invasive breast cancer cells [
140]. Inhibition of Akt also downregulates Twist in cancer cells [
80]. Furthermore, Akt phosphorylates and activates Twist1, which in turn enhances the phosphorylation of Akt because of increased TGFβ signalling in human breast cancer [
141,
142,
143]. Data also suggest that the polycomb group protein named B lymphoma Mo-MLV insertion region 1 homolog (Bmi1) is a downstream target of Twist1 and is crucial for EMT and cancer metastasis [
144]. Akt can phosphorylate Bmi1 directly in high-grade prostate tumours [
145]. Promotion of Akt activity by Bmi1 was also found to promote EMT by blocking the GSK3β-mediated degradation of Snail in HNSCC and breast cancer [
146,
147].
4. Akt in HNSCC Metastasis
Head and neck squamous cell carcinoma (HNSCC) denotes epithelial tumours that develop in the oral cavity, pharynx, larynx, and nasal cavity. The main risk factors of HNSCC are alcohol and tobacco use and HPV infection [
153,
154]. It is the seventh most common cancer worldwide, with more than 887,000 cases and 450,000 deaths every year (accumulation of different head and neck cancer sites) [
155]. It has recently been shown that Akt is persistently activated in the vast majority of HNSCC cases. Active forms of Akt (phosphorylated) can readily be detected in both experimental and human HNSCCs and HNSCC-derived cell lines [
156,
157,
158]. Akt can be phosphorylated, hence activated by different growth factors, chemokines, integrins, etc., and their respective receptors, ras mutations, PI3Ka gene amplification, overexpression, or activating mutations. Akt can also be activated by aberrant PTEN activity due to genetic alterations or reduced expression in HNSCC [
159,
160]. Akt activation is an early event in HNSCC progression which can be identified in as many as 50% of tongue preneoplastic lesions [
161]. Akt activation also represents an independent prognostic marker of poor clinical outcome in both tongue and oropharyngeal HNSCCs [
161,
162] and is linked with the conversion of a potentially malignant oral lesion to OSCC (oral squamous cell carcinoma) [
163].
Akt is known to induce morphological changes associated with EMT, loss of cell–cell adhesion, and increased motility and invasion in HNSCC [
112]. Oral carcinoma cells, of epithelial origin, ectopically express a mesenchyme-specific transcription factor (HMGA2) at the invasive front, which has a significant impact on tumour progression and patient survival [
164]. However, the definitive evidence that EMT was induced by Akt was provided by a study in which oral squamous cell carcinoma cell lines overexpressing activated mutant Akt were shown to undergo EMT and downregulate E-cadherin [
127]. Snail and SIP1 exhibit an inverse correlation with E-cadherin expression levels in oral carcinoma cells [
165,
166]. An OSCC clone with stable Snail overexpression displayed spindle morphology, amplified expression of vimentin, and reduced expression of E-cadherin [
167].
5. Conclusions
Extensive studies have demonstrated that the activation of Akt by phosphorylation of different amino acid residues determines substrate selectivity and thus exerts different biological activity in different cell types. Three highly homologous Akt isoforms have non-overlapping and opposing functions in different cancer types. As Akt is the central signalling node that incorporates cell membrane, cytoplasmic and nuclear signals regulating cell fate, analysing Akt isoforms and cell-type-specific signalling pathways and targeting them will contribute to personalised targeted HNSCC therapy. Thus, carefully designing a clinical study using a combination of a PI3K-Akt pathway inhibitor and another signalling molecule inhibitor or receptor inhibitor during the early stages of HNSCC might result in an expected positive outcome.