 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Robert Vink | -- | 3530 | 2023-11-15 00:48:08 | | | |
| 2 | Camila Xu | Meta information modification | 3530 | 2023-11-15 02:27:49 | | |
Video Upload Options
Traumatic brain injury (TBI) is an acquired insult to the brain caused by external mechanical impact and/or acceleration forces that result in transient or permanent neurological dysfunction. Substance P is a member of the tachykinin protein family whose neuronal release after TBI plays a critical role in TBI pathophysiology, including the development of post-traumatic oedema, increased intracranial pressure, neuroinflammation, neuronal cell death, and neurodegeneration. Because substance P release after TBI is dependent on the intensity and frequency of injury-related mechanical stimulation, the degree and anatomical distribution of substance P receptor activation after TBI will vary with injury severity and frequency, resulting in different outcomes for different injuries.
1. Introduction
2. Mild TBI
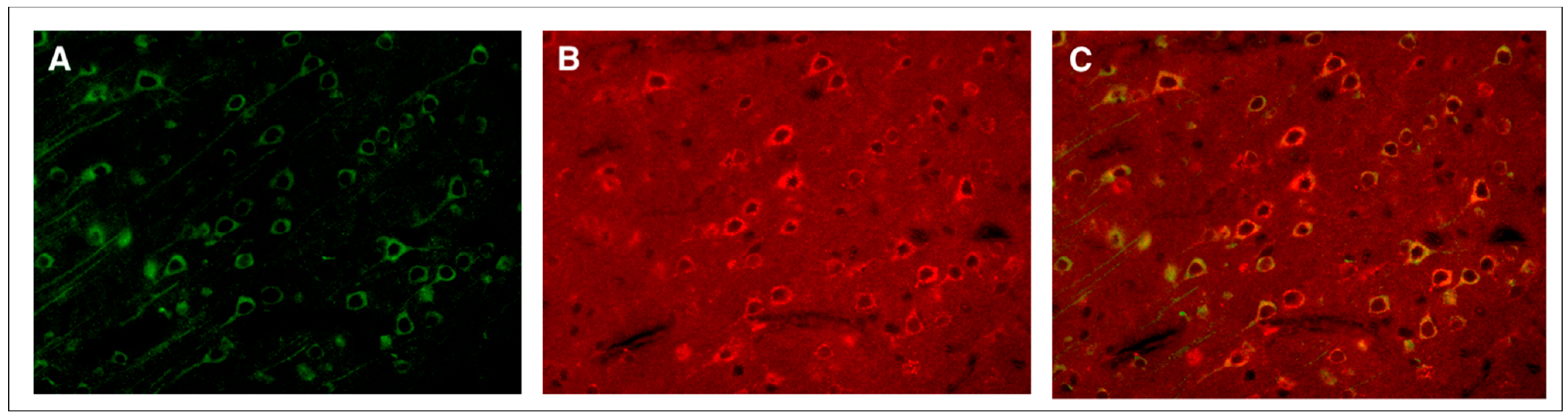
3. Moderate TBI
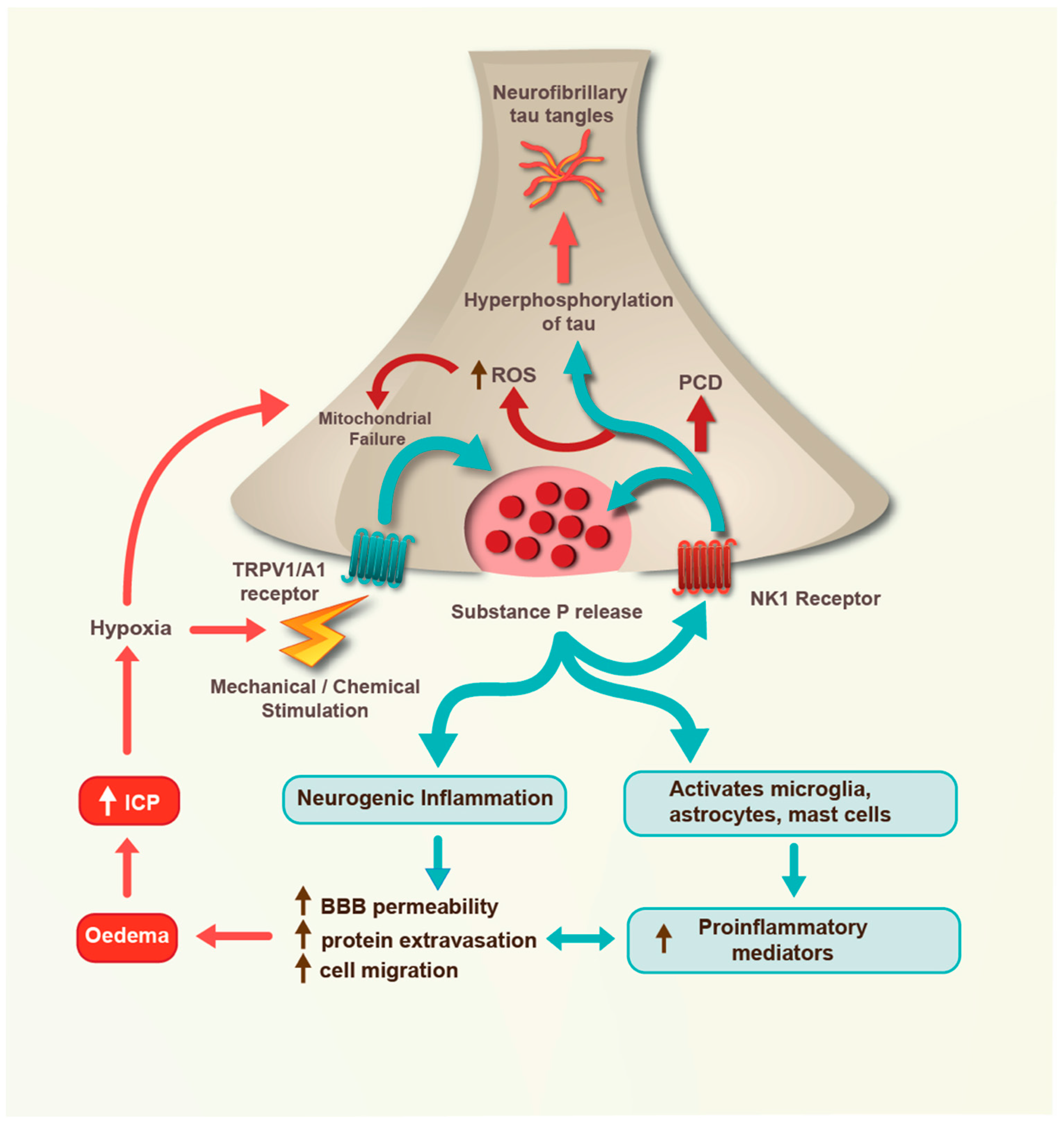
4. Severe TBI
References
- Centers for Disease Control and Prevention. Leading Causes of Death and Injury. Available online: https://www.cdc.gov/injury/wisqars/leadingcauses.html (accessed on 25 June 2023).
- Goldstein, M. Traumatic brain injury: A silent epidemic. Ann. Neurol. 1990, 27, 327.
- Nowinski, C.J.; Bureau, S.C.; Buckland, M.E.; Curtis, M.A.; Daneshvar, D.H.; Faull, R.L.M.; Grinberg, L.T.; Hill-Yardin, E.L.; Murray, H.C.; Pearce, A.J.; et al. Applying the Bradford Hill criteria for causation to repetitive head impacts and chronic traumatic tncephalopathy. Front. Neurol. 2022, 13, 938163.
- Stocchetti, N.; Carbonara, M.; Citerio, G.; Ercole, A.; Skrifvars, M.B.; Smielewski, P.; Zoerle, T.; Menon, D.K. Severe traumatic brain injury: Targeted management in the intensive care unit. Lancet Neurol. 2017, 16, 452–464.
- Hokfelt, T.; Pernow, B.; Wahren, J. Substance P: A pioneer amongst neuropeptides. J. Intern. Med. 2001, 249, 27–40.
- Coveñas, R.; Rodriguez, F.D.; Muñoz, M. The Neurokinin-1 receptor: A promising antitumor target. Receptors 2022, 1, 72–97.
- Almeida, T.A.; Rojo, J.; Nieto, P.M.; Pinto, F.M.; Hernandez, M.; Martin, J.D.; Candenas, M.L. Tachykinins and tachykinin receptors: Structure and activity relationships. Curr. Med. Chem. 2004, 11, 2045–2081.
- Saria, A. The tachykinin NK1 receptor in the brain: Pharmacology and putative functions. Eur. J. Pharmacol. 1999, 375, 51–60.
- Iverson, G.L.; Gardner, A.J.; Terry, D.P.; Ponsford, J.L.; Sills, A.K.; Broshek, D.K.; Solomon, G.S. Predictors of clinical recovery from concussion: A systematic review. Br. J. Sports Med. 2017, 51, 941–948.
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.S.; Kubilus, C.A.; Stern, R.A. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735.
- Donkin, J.J.; Cernak, I.; Rodgers, K.M.; Vink, R. Mild concussive head injury results in increased brain substance P immunoreactivity. In 7th International Neurotrauma Symposium; Medimond International Proceedings: Bologna, Italy, 2004; pp. 75–78.
- Campos, M.M.; Calixto, J.B. Neurokinin mediation of edema and inflammation. Neuropeptides 2000, 34, 314–322.
- McKee, A.C.; Stein, T.D.; Kiernan, P.T.; Alvarez, V.E. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 2015, 25, 350–364.
- McKee, A.C.; Stein, T.D.; Huber, B.R.; Crary, J.F.; Bieniek, K.; Dickson, D.; Alvarez, V.E.; Cherry, J.D.; Farrell, K.; Butler, M.; et al. Chronic traumatic encephalopathy (CTE): Criteria for neuropathological diagnosis and relationship to repetitive head impacts. Acta Neuropathol. 2023, 145, 371–394.
- O’Keeffe, E.; Kelly, E.; Liu, Y.; Giordano, C.; Wallace, E.; Hynes, M.; Tiernan, S.; Meagher, A.; Greene, C.; Hughes, S.; et al. Dynamic blood-brain barrier regulation in mild traumatic brain injury. J. Neurotrauma 2020, 37, 347–356.
- Corrigan, F.; Cernak, I.; McAteer, K.; Hellewell, S.C.; Rosenfeld, J.V.; Turner, R.J.; Vink, R. NK1 antagonists attenuate tau phosphorylation after blast and repeated concussive injury. Sci. Rep. 2021, 11, 8861.
- Hoffmann, T.; Nimmo, A.J.; Sleight, A.; Vankan, P.; Vink, R. Use of NK-1 Receptor Antagonists with Pyridinic Structure, for the Treatment of Brain, Spinal or Nerve Injury. WO2003006016A3, 2003.
- Fiebich, B.L.; Schleicher, S.; Butcher, R.D.; Craig, A.; Lieb, K. The neuropeptide substance P activates p38 mitogen-activated protein kinase resulting in IL-6 expression independently from NF-kappa B. J. Immunol. 2000, 165, 5606–5611.
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119.
- Sun, J.; Ramnath, R.D.; Tamizhselvi, R.; Bhatia, M. Role of protein kinase C and phosphoinositide 3-kinase-Akt in substance P-induced proinflammatory pathways in mouse macrophages. FASEB J. 2009, 23, 997–1010.
- Mantyh, P.W. Neurobiology of substance P and the NK1 receptor. J. Clin. Psychiatry 2002, 63, 6–10.
- Collins-Praino, L.; Gutschmidt, D.; Sharkey, J.; Arulsamy, A.; Corrigan, F. Temporal changes in tau phosphorylation and related kinase and phosphatases following two models of traumatic brain injury. J. Neuroinflamm. Neurodegener. Dis. 2018, 2, 100007.
- Murphy, J.E.; Roosterman, D.; Cottrell, G.S.; Padilla, B.E.; Feld, M.; Brand, E.; Cedron, W.J.; Bunnett, N.W.; Steinhoff, M. Protein phosphatase 2A mediates resensitization of the neurokinin 1 receptor. Am. J. Physiol. Cell Physiol. 2011, 301, C780–C791.
- Nimmo, A.J.; Cernak, I.; Heath, D.L.; Hu, X.; Bennett, C.J.; Vink, R. Neurogenic inflammation is associated with development of edema and functional deficits following traumatic brain injury in rats. Neuropeptides 2004, 38, 40–47.
- Vink, R.; Young, A.; Bennett, C.J.; Hu, X.; Connor, C.O.; Cernak, I.; Nimmo, A.J. Neuropeptide release influences brain edema formation after diffuse traumatic brain injury. Acta Neurochir. Suppl. 2003, 86, 257–260.
- Corrigan, F.; Leonard, A.; Ghabriel, M.; Van Den Heuvel, C.; Vink, R. A substance P antagonist improves outcome in female Sprague Dawley rats following diffuse traumatic brain injury. CNS Neurosci. Ther. 2012, 18, 513–515.
- Donkin, J.J.; Cernak, I.; Blumberg, P.C.; Vink, R. A substance P antagonist reduces axonal injury and improves neurologic outcome when administered up to 12 hours after traumatic brain injury. J. Neurotrauma 2011, 28, 218–224.
- Donkin, J.J.; Nimmo, A.J.; Cernak, I.; Blumbergs, P.C.; Vink, R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J. Cereb. Blood Flow. Metab. 2009, 29, 1388–1398.
- Zacest, A.C.; Vink, R.; Manavis, J.; Sarvestani, G.T.; Blumbergs, P.C. Substance P immunoreactivity increases following human traumatic brain injury. Acta Neurochir. Suppl. 2010, 106, 211–216.
- Donkin, J.J.; Vink, R. Mechanisms of cerebral edema in traumatic brain injury: Therapeutic developments. Curr. Opin. Neurol. 2010, 23, 293–299.
- Hooper, C.; Pinteaux-Jones, F.; Fry, V.A.; Sevastou, I.G.; Baker, D.; Heales, S.J.; Pocock, J.M. Differential effects of albumin on microglia and macrophages; implications for neurodegeneration following blood-brain barrier damage. J. Neurochem. 2009, 109, 694–705.
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflammation 2016, 13, 264.
- Safwat, A.; Helmy, A.; Gupta, A. The role of substance P within traumatic brain injury and implications for therapy. J. Neurotrauma 2023. Online ahead of print.
- Seabrook, G.R.; Shepheard, S.L.; Williamson, D.J.; Tyrer, P.; Rigby, M.; Cascieri, M.A.; Harrison, T.; Hargreaves, R.J.; Hill, R.G. L-733,060, a novel tachykinin NK1 receptor antagonist; effects in i mobilisation, cardiovascular and dural extravasation assays. Eur. J. Pharmacol. 1996, 317, 129–135.
- Sirianni, A.C.; Jiang, J.; Zeng, J.; Mao, L.L.; Zhou, S.; Sugarbaker, P.; Zhang, X.; Li, W.; Friedlander, R.M.; Wang, X. N-acetyl-l-tryptophan, but not N-acetyl-d-tryptophan, rescues neuronal cell death in models of amyotrophic lateral sclerosis. J. Neurochem. 2015, 134, 956–968.
- Matalinska, J.; Lipinski, P.F.J. Correcting a widespread error: Neuroprotectant N-acetyl-L-tryptophan does not bind to the neurokinin-1 receptor. Mol. Cell Neurosci. 2022, 120, 103728.
- Corrigan, F.; Vink, R.; Turner, R.J. Inflammation in acute CNS injury: A focus on the role of substance P. Br. J. Pharmacol. 2016, 173, 703–715.
- Li, Q.; Wu, X.; Yang, Y.; Zhang, Y.; He, F.; Xu, X.; Zhang, Z.; Tao, L.; Luo, C. Tachykinin NK1 receptor antagonist L-733,060 and substance P deletion exert neuroprotection through inhibiting oxidative stress and cell death after traumatic brain injury in mice. Int. J. Biochem. Cell Biol. 2019, 107, 154–165.
- Habgood, M.D.; Bye, N.; Dziegielewska, K.M.; Ek, C.J.; Lane, M.A.; Potter, A.; Morganti-Kossmann, C.; Saunders, N.R. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur. J. Neurosci. 2007, 25, 231–238.
- Castro-Obregón, S.; Del Rio, G.; Chen, S.F.; Swanson, R.A.; Frankowski, H.; Rao, R.V.; Stoka, V.; Vesce, S.; Nicholls, D.G.; Bredesen, D.E. A ligand-receptor pair that triggers a non-apoptotic form of programmed cell death. Cell. Death Differ. 2002, 9, 807–817.
- Vink, R.; Donkin, J.J.; Cruz, M.I.; Nimmo, A.J.; Cernak, I. A substance P antagonist increases brain intracellular free magnesium concentration after diffuse traumatic brain injury in rats. J. Am. Coll. Nutr. 2004, 23, 538S–540S.
- Lorente, L.; Martín, M.M.; Almeida, T.; Hernandez, M.; Ramos, L.; Argueso, M.; Cáceres, J.J.; Solé-Violán, J.; Jiménez, A. Serum substance P levels are associated with severity and mortality in patients with severe traumatic brain injury. Crit. Care 2015, 19, 192.
- Lorente, L.; Martín, M.M.; Pérez-Cejas, A.; González-Rivero, A.F.; Argueso, M.; Ramos, L.; Solé-Violán, J.; Cáceres, J.J.; Jiménez, A.; García-Marin, V. Persistently high serum substance P levels and early mortality in patients with severe traumatic brain injury. World Neurosurg. 2019, 132, e613–e617.
- Zhou, Y.; Ye, H.; Lu, W. Serum substance P concentration in children with traumatic brain injury: A First Report. World Neurosurg. 2021, 147, e200–e205.
- Alves, J.L.; Mendes, J.; Leitao, R.; Silva, A.P.; Pinto, A.M. A multi-staged neuropeptide response to traumatic brain injury. Eur. J. Trauma Emerg. Surg. 2022, 48, 507–517.
- Gabrielian, L.; Wisllshire, L.W.; Helps, S.C.; van den Heuvel, C.; Mathias, J.L.; Vink, R. Intracranial pressure changes following traumatic brain injury in rats: Lack of significant change in the absence of mass lesions or hypoxia. J. Neurotrauma 2011, 28, 2103–2111.
- Vink, R.; Bahtia, K.D.; Reilly, P.L. The relationship between intracranial pressure and brain oxygenation following traumatic brain injury in sheep. Acta Neurochir. Suppl. 2008, 102, 189–192.
- Gabrielian, L.; Helps, S.C.; Thornton, E.; Turner, R.J.; Leonard, A.V.; Vink, R. Substance P antagonists as a novel intervention for brain edema and raised intracranial pressure. Acta Neurochir. Suppl. 2013, 118, 201–204.
- Vink, R.; Gabrielian, L.; Thornton, E. The role of substance P in secondary pathophysiology after traumatic brain injury. Front. Neurol. 2017, 8, 304.
- O’Connor, C.A.; Cernak, I.; Vink, R. The temporal profile of edema formation differs between male and female rats following diffuse traumatic brain injury. Acta Neurochir. Suppl. 2006, 96, 121–124.
- Sorby-Adams, A.J.; Leonard, A.V.; Hoving, J.W.; Yassi, N.; Vink, R.; Wells, A.J.; Turner, R.J. NK1-r antagonist treatment comparable to decompressive craniectomy in reducing intracranial pressure following stroke. Front. Neurosci. 2019, 13, 681.
- Nag, S.; Venugopalan, R.; Stewart, D.J. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 2007, 114, 459–469.
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53.
- Schubert, W.; Frank, P.G.; Razani, B.; Park, D.S.; Chow, C.W.; Lisanti, M.P. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J. Biol. Chem. 2001, 276, 48619–48622.
- Povlishock, J.T.; Becker, D.P.; Sullivan, H.G.; Miller, J.D. Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain Res. 1978, 153, 223–239.
- Barzo, P.; Marmarou, A.; Fatouros, P.; Corwin, F.; Dunbar, J.G. Acute blood-brain barrier changes in experimental closed head injury as measured by MRI and Gd-DTPA. Acta Neurochir. Suppl. 1997, 70, 243–246.
- Castejón, O.J. Increased vesicular and vacuolar transendothelial transport in traumatic human brain oedema. A review. Folia Neuropathol. 2013, 51, 93–102.
- Kubale, V.; Abramovic, Z.; Pogacnik, A.; Heding, A.; Sentjurc, M.; Vrecl, M. Evidence for a role of caveolin-1 in neurokinin-1 receptor plasma-membrane localization, efficient signaling, and interaction with beta-arrestin 2. Cell Tissue Res. 2007, 330, 231–245.
- Meyer, B.H.; Segura, J.M.; Martinez, K.L.; Hovius, R.; George, N.; Johnsson, K.; Vogel, H. FRET imaging reveals that functional neurokinin-1 receptors are monomeric and reside in membrane microdomains of live cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2138–2143.
- Monastyrskaya, K.; Hostettler, A.; Buergi, S.; Draeger, A. The NK1 receptor localizes to the plasma membrane microdomains, and its activation is dependent on lipid raft integrity. J. Biol. Chem. 2005, 280, 7135–7146.
- Mineo, C.; Ying, Y.S.; Chapline, C.; Jaken, S.; Anderson, R.G. Targeting of protein kinase Calpha to caveolae. J. Cell Biol. 1998, 141, 601–610.

