 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ali Mussa | -- | 5034 | 2023-11-13 10:03:03 | | | |
| 2 | Peter Tang | + 1 word(s) | 5035 | 2023-11-13 10:06:59 | | |
Video Upload Options
The idea that Vitamin C (Vit-C) could be utilized as a form of anti-cancer therapy has generated many contradictory arguments. Insights into the physiological characteristics of Vit-C, its pharmacokinetics, and results from preclinical reports, however, suggest that high-dose Vit-C could be effectively utilized in the management of various tumor types. Studies have shown that the pharmacological action of Vit-C can attack various processes that cancerous cells use for their growth and development.
1. Introduction
2. Historical Background and Justification of High-Dose Vitamin-C in Cancer Management
3. Different Oxidized Forms of Vitamin-C
4. Enzymatic Activities of Vitamin-C
5. ROS
6. Anti-Cancer Mechanisms of Vitamin-C
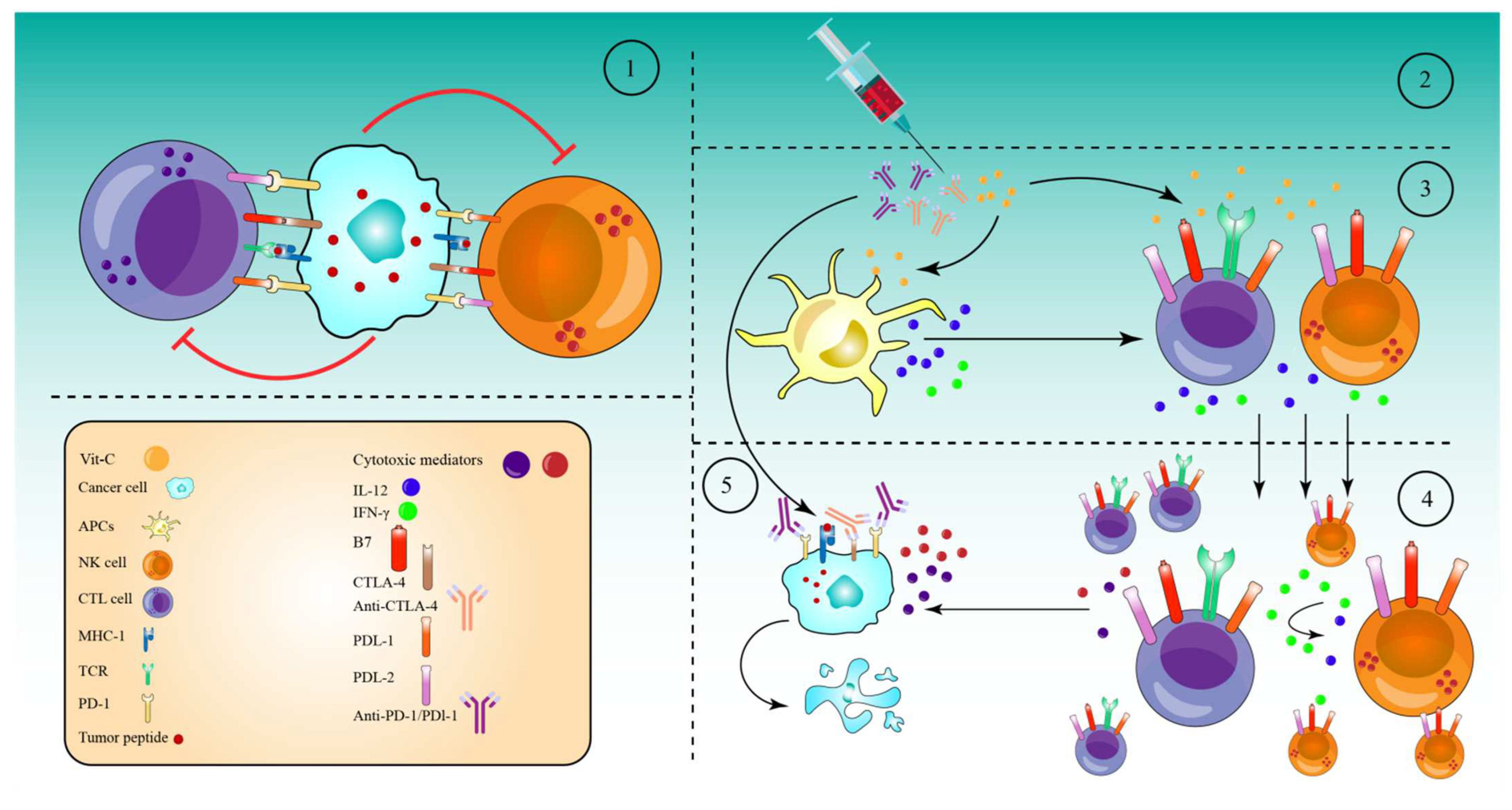
7. High-Dose Vitamin-C Enhances Cancer Immunotherapy
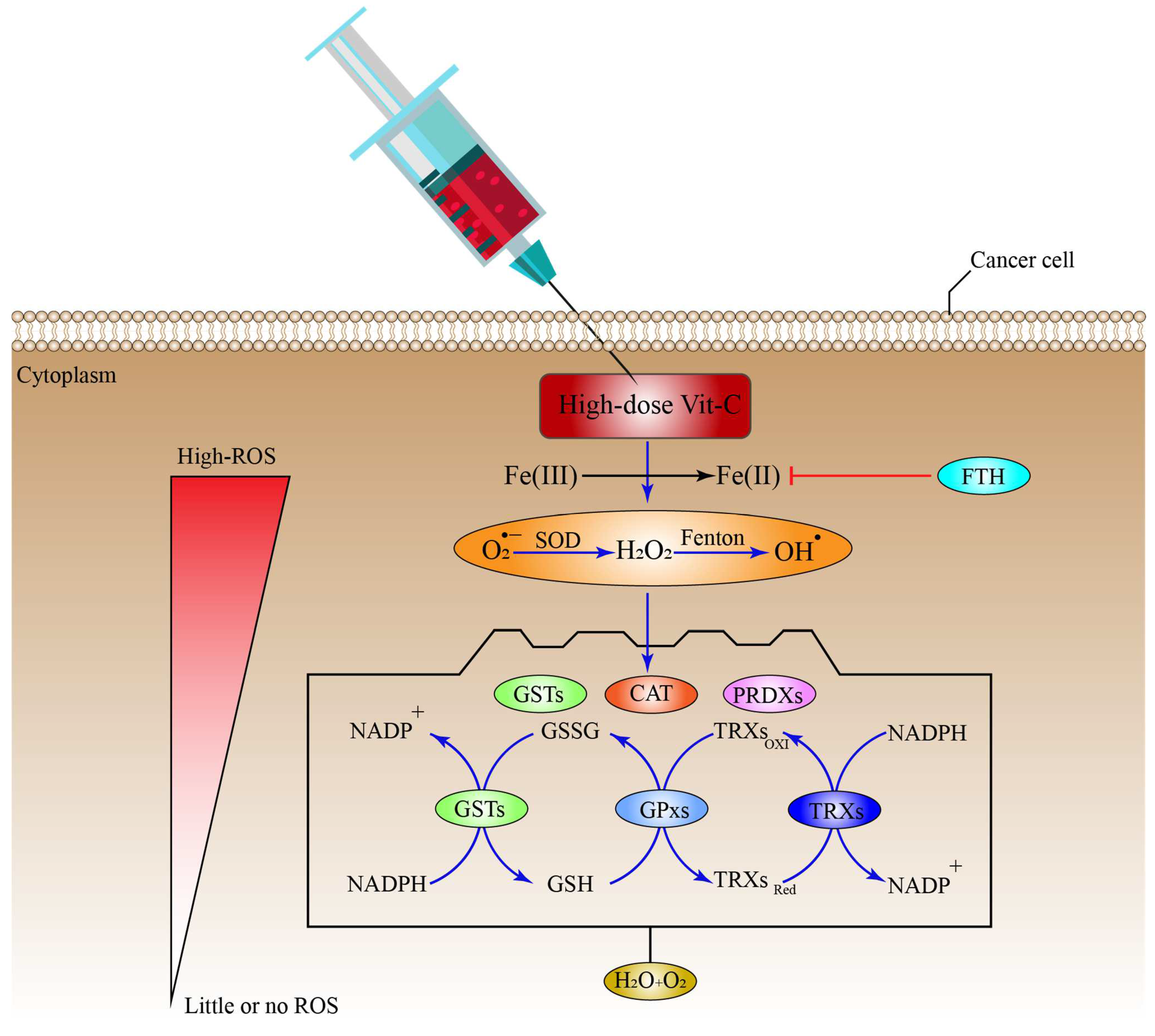
8. Antioxidant Systems That Might Inhibit the Effect of High-Dose Vitamin-C Therapy
|
Defense System |
Sub-Cellular Localization(s) |
ROS Type and System Function |
Ref. |
|---|---|---|---|
|
SODs |
Cytosol and Peroxisomes |
O2•− to O2 and H2O2 |
[80] |
|
CAT |
Peroxisomes |
H2O2 to H2O and O2 |
[80] |
|
GPxs |
Mitochondria and Cytosol |
H2O2 to H2O lipid peroxides to alcohols |
[80] |
|
PRDXs |
Cytosol, Mitochondria, Nucleus And Endoplasmic reticulum |
H2O2 to H2O |
[95] |
|
TRXs |
Cytosol, Mitochondria, Nucleus |
H2O2 to H2O and O2 |
|
|
GSH |
Cytosol, Mitochondria, Nucleus And Endoplasmic reticulum |
H2O2 to H2O |
|
|
GSTs * |
Cytosol, Membrane-bound |
conjugates with reduced GSH) |
[110] |
|
FTH * |
Nucleus, Lysosome, Cytoplasm |
Sequester Fe(II) to inhibit ROS generation |
[112] |
SODs, superoxide dismutases; CAT, catalase; PRDXs, Peroxiredoxins; GSTs, glutathione-S-transferases; TRXs, Thioredoxins; GPxs, glutathione peroxidases; GSH, reduced glutathione; GSSG; FTH, ferritin heavy chain, indirect ROS formation inhibition *.
References
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Yagi, K. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-gamma-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 13685–13688.
- Drouin, G.; Godin, J.-R.; Pagé, B. The genetics of vitamin C loss in vertebrates. Curr. Genom. 2011, 12, 371–378.
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396.
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1999, 13, 1007–1024.
- Englard, S.; Seifter, S. The biochemical functions of ascorbic acid. Annu. Rev. Nutr. 1986, 6, 365–406.
- Kuiper, C.; Vissers, M.C.M. Ascorbate as a Co-Factor for Fe- and 2-Oxoglutarate Dependent Dioxygenases: Physiological Activity in Tumor Growth and Progression. Front. Oncol. 2014, 4, 359.
- Young, J.I.; Züchner, S.; Wang, G. Regulation of the Epigenome by Vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564.
- Carr, A.C.; McCall, C. The role of vitamin C in the treatment of pain: New insights. J. Transl. Med. 2017, 15, 77.
- Ting, H.H.; Timimi, F.K.; Boles, K.S.; Creager, S.J.; Ganz, P.; Creager, M.A. Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1996, 97, 22–28.
- Salonen, R.M.; Nyyssönen, K.; Kaikkonen, J.; Porkkala-Sarataho, E.; Voutilainen, S.; Rissanen, T.H.; Tuomainen, T.P.; Valkonen, V.P.; Ristonmaa, U.; Lakka, H.M.; et al. Six-year effect of combined vitamin C and E supplementation on atherosclerotic progression: The Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) Study. Circulation 2003, 107, 947–953.
- Van Straten, M.; Josling, P. Preventing the common cold with a vitamin C supplement: A double-blind, placebo-controlled survey. Adv. Ther. 2002, 19, 151–159.
- Valero, M.P.; Fletcher, A.E.; De Stavola, B.L.; Vioque, J.; Alepuz, V.C. Vitamin C is associated with reduced risk of cataract in a Mediterranean population. J. Nutr. 2002, 132, 1299–1306.
- Ramdas, W.D.; Schouten, J.; Webers, C.A.B. The Effect of Vitamins on Glaucoma: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 359.
- Seddon, J.M.; Ajani, U.A.; Sperduto, R.D.; Hiller, R.; Blair, N.; Burton, T.C.; Farber, M.D.; Gragoudas, E.S.; Haller, J.; Miller, D.T.; et al. Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group. Jama 1994, 272, 1413–1420.
- Chen, G.C.; Lu, D.B.; Pang, Z.; Liu, Q.F. Vitamin C intake, circulating vitamin C and risk of stroke: A meta-analysis of prospective studies. J. Am. Heart Assoc. 2013, 2, e000329.
- Losonczy, K.G.; Harris, T.B.; Havlik, R.J. Vitamin E and vitamin C supplement use and risk of all-cause and coronary heart disease mortality in older persons: The Established Populations for Epidemiologic Studies of the Elderly. Am. J. Clin. Nutr. 1996, 64, 190–196.
- Zhang, J.; Rao, X.; Li, Y.; Zhu, Y.; Liu, F.; Guo, G.; Luo, G.; Meng, Z.; De Backer, D.; Xiang, H.; et al. Pilot trial of high-dose vitamin C in critically ill COVID-19 patients. Ann. Intensive Care 2021, 11, 5.
- Cameron, E.; Campbell, A. The orthomolecular treatment of cancer. II. Clinical trial of high-dose ascorbic acid supplements in advanced human cancer. Chem. Biol. Interact. 1974, 9, 285–315.
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 3685–3689.
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Reevaluation of prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1978, 75, 4538–4542.
- Murata, A.; Morishige, F.; Yamaguchi, H. Prolongation of survival times of terminal cancer patients by administration of large doses of ascorbate. Int. J. Vitam. Nutr. Res. 1982, 23, 103–113.
- Creagan, E.T.; Moertel, C.G.; O’Fallon, J.R.; Schutt, A.J.; O’Connell, M.J.; Rubin, J.; Frytak, S. Failure of High-Dose Vitamin C (Ascorbic Acid) Therapy to Benefit Patients with Advanced Cancer. N. Engl. J. Med. 1979, 301, 687–690.
- Moertel, C.G.; Fleming, T.R.; Creagan, E.T.; Rubin, J.; O’Connell, M.J.; Ames, M.M. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N. Engl. J. Med. 1985, 312, 137–141.
- Block, G. Vitamin C and cancer prevention: The epidemiologic evidence. Am. J. Clin. Nutr. 1991, 53 (Suppl. 1), 270s–282s.
- Block, G. Epidemiologic evidence regarding vitamin C and cancer. Am. J. Clin. Nutr. 1991, 54 (Suppl. 6), 1310s–1314s.
- Head, K.A. Ascorbic acid in the prevention and treatment of cancer. Altern. Med. Rev. A J. Clin. Ther. 1998, 3, 174–186.
- Block, G.; Patterson, B.; Subar, A. Fruit, vegetables, and cancer prevention: A review of the epidemiological evidence. Nutr. Cancer 1992, 18, 1–29.
- Steinmetz, K.A.; Potter, J.D. Vegetables, fruit, and cancer prevention: A review. J. Am. Diet. Assoc. 1996, 96, 1027–1039.
- Loria, C.M.; Klag, M.J.; Caulfield, L.E.; Whelton, P.K. Vitamin C status and mortality in US adults. Am. J. Clin. Nutr. 2000, 72, 139–145.
- Padayatty, S.J.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Hoffer, L.J.; Levine, M. Intravenously administered vitamin C as cancer therapy: Three cases. CMAJ 2006, 174, 937–942.
- Dachs, G.U.; Gandhi, J.; Wohlrab, C.; Carr, A.C.; Morrin, H.R.; Pullar, J.M.; Bayer, S.B.; Eglinton, T.W.; Robinson, B.A.; Vissers, M.C.M. Vitamin C Administration by Intravenous Infusion Increases Tumor Ascorbate Content in Patients With Colon Cancer: A Clinical Intervention Study. Front. Oncol. 2021, 10, 600715.
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609.
- Cabanillas, F. Vitamin C and cancer: What can we conclude—1609 patients and 33 years later? Puerto Rico Health Sci. J. 2010, 29, 215–217.
- Cha, J.; Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Ascorbate depletion increases growth and metastasis of melanoma cells in vitamin C deficient mice. Exp. Oncol. 2011, 33, 226–230.
- Cha, J.; Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Ascorbate supplementation inhibits growth and metastasis of B16FO melanoma and 4T1 breast cancer cells in vitamin C-deficient mice. Int. J. Oncol. 2013, 42, 55–64.
- Magrì, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chilà, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707.
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537.
- Kushi, L.H.; Fee, R.M.; Sellers, T.A.; Zheng, W.; Folsom, A.R. Intake of vitamins A, C, and E and postmenopausal breast cancer. The Iowa Women’s Health Study. Am. J. Epidemiol. 1996, 144, 165–174.
- Roa, F.J.; Pena, E.; Gatica, M.; Escobar-Acuna, K.; Saavedra, P.; Maldonado, M.; Cuevas, M.; Moraga-Cid, G.; Rivas, C.I.; Munoz-Montesino, C. Therapeutic Use of Vitamin C in Cancer: Physiological Considerations. Front. Pharmacol. 2020, 11, 211.
- Assouline, S.; Miller, W.H. High-dose vitamin C therapy: Renewed hope or false promise? CMAJ 2006, 174, 956–957.
- Yeom, C.H.; Jung, G.C.; Song, K.J. Changes of terminal cancer patients’ health-related quality of life after high dose vitamin C administration. J. Korean Med. Sci. 2007, 22, 7–11.
- Corti, A.; Casini, A.F.; Pompella, A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch. Biochem. Biophys. 2010, 500, 107–115.
- Glorieux, C.; Buc Calderon, P. Vitamin C (Ascorbate) and Redox Topics in Cancer. Antioxid Redox Signal 2021, 35, 1157–1175.
- Vera, J.C.; Rivas, C.I.; Zhang, R.H.; Farber, C.M.; Golde, D.W. Human HL-60 myeloid leukemia cells transport dehydroascorbic acid via the glucose transporters and accumulate reduced ascorbic acid. Blood 1994, 84, 1628–1634.
- Padayatty, S.J.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral Dis. 2016, 22, 463–493.
- Wilson, J.X. The physiological role of dehydroascorbic acid. FEBS Lett. 2002, 527, 5–9.
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; Del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965.
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543.
- Podmore, I.D.; Griffiths, H.R.; Herbert, K.E.; Mistry, N.; Mistry, P.; Lunec, J. Vitamin C exhibits pro-oxidant properties. Nature 1998, 392, 559.
- Flashman, E.; Davies, S.L.; Yeoh, K.K.; Schofield, C.J. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem. J. 2010, 427, 135–142.
- Dao, J.H.; Kurzeja, R.J.; Morachis, J.M.; Veith, H.; Lewis, J.; Yu, V.; Tegley, C.M.; Tagari, P. Kinetic characterization and identification of a novel inhibitor of hypoxia-inducible factor prolyl hydroxylase 2 using a time-resolved fluorescence resonance energy transfer-based assay technology. Anal. Biochem. 2009, 384, 213–223.
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free. Radic. Biol. Med. 2015, 82, 22–28.
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824.
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175.
- Muthukumar, K.; Nachiappan, V. Cadmium-induced oxidative stress in Saccharomyces cerevisiae. Indian J. Biochem. Biophys. 2010, 47, 383–387.
- Tominaga, H.; Kodama, S.; Matsuda, N.; Suzuki, K.; Watanabe, M. Involvement of reactive oxygen species (ROS) in the induction of genetic instability by radiation. J. Radiat. Res. 2004, 45, 181–188.
- van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.L.; Gans, R.O.B.; Koëter, G.H.; Choi, A.M.K.; van Oosterhout, A.J.M.; Slebos, D.-J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung. Cell Mol. Physiol. 2009, 297, L109–L114.
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-Induced Oxidative Stress and Toxicity. J. Toxicol. 2012, 2012, 645460.
- Klotz, L.-O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654.
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511.
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free. Radic Res. 2010, 44, 479–496.
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176.
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. CB 2014, 24, R453–R462.
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429.
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735.
- Thyagarajan, A.; Sahu, R.P. Potential Contributions of Antioxidants to Cancer Therapy: Immunomodulation and Radiosensitization. Integr. Cancer Ther. 2018, 17, 210–216.
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra215.
- Zou, Z.V.; Le Gal, K.; El Zowalaty, A.E.; Pehlivanoglu, L.E.; Garellick, V.; Gul, N.; Ibrahim, M.X.; Bergh, P.-O.; Henricsson, M.; Wiel, C.; et al. Antioxidants Promote Intestinal Tumor Progression in Mice. Antioxidants 2021, 10, 241.
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556.
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L.; Valanis, B.; Williams, J.H.; et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N. Engl. J. Med. 1996, 334, 1150–1155.
- Wondrak, G.T. Redox-directed cancer therapeutics: Molecular mechanisms and opportunities. Antioxid Redox Signal 2009, 11, 3013–3069.
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11.
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255.
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16.
- Luchtel, R.A.; Bhagat, T.; Pradhan, K.; Jacobs, W.R.; Levine, M.; Verma, A.; Shenoy, N. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl. Acad. Sci. USA 2020, 117, 1666–1677.
- Peng, D.; He, A.; He, S.; Ge, G.; Wang, S.; Ci, W.; Li, X.; Xia, D.; Zhou, L. Ascorbic acid induced TET2 enzyme activation enhances cancer immunotherapy efficacy in renal cell carcinoma. Int. J. Biol. Sci. 2022, 18, 995–1007.
- Xu, Y.-p.; Lv, L.; Liu, Y.; Smith, M.D.; Li, W.-C.; Tan, X.-m.; Cheng, M.; Li, Z.; Bovino, M.; Aubé, J.; et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Investig. 2019, 129, 4316–4331.
- Ma, J.; Zhang, C.; Shi, G.; Yue, D.; Shu, Y.; Hu, S.; Qi, Z.; Chen, Y.; Zhang, B.; Zhang, Y.; et al. High-dose VitC plus oncolytic adenoviruses enhance immunogenic tumor cell death and reprogram tumor immune microenvironment. Mol. Ther. 2022, 30, 644–661.
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293.
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153.
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297.
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin Heavy Chain Upregulation by NF-κB Inhibits TNFα-Induced Apoptosis by Suppressing Reactive Oxygen Species. Cell 2004, 119, 529–542.
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93.
- Ekoue, D.N.; He, C.; Diamond, A.M.; Bonini, M.G. Manganese superoxide dismutase and glutathione peroxidase-1 contribute to the rise and fall of mitochondrial reactive oxygen species which drive oncogenesis. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1858, 628–632.
- Dhar, S.K.; St. Clair, D.K. Manganese superoxide dismutase regulation and cancer. Free. Radic. Biol. Med. 2012, 52, 2209–2222.
- Becuwe, P.; Ennen, M.; Klotz, R.; Barbieux, C.; Grandemange, S. Manganese superoxide dismutase in breast cancer: From molecular mechanisms of gene regulation to biological and clinical significance. Free. Radic. Biol. Med. 2014, 77, 139–151.
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090.
- Bauer, G. Tumor Cell-protective Catalase as a Novel Target for Rational Therapeutic Approaches Based on Specific Intercellular ROS Signaling. Anticancer. Res. 2012, 32, 2599–2624.
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108.
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997.
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3289–3303.
- Liu, J.; Hinkhouse, M.M.; Sun, W.; Weydert, C.J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. GoswamiRedox Regulation of Pancreatic Cancer Cell Growth: Role of Glutathione Peroxidase in the Suppression of the Malignant Phenotype. Hum. Gene Ther. 2004, 15, 239–250.
- Jardim, B.V.; Moschetta, M.G.; Leonel, C.; Gelaleti, G.B.; Regiani, V.R.; Ferreira, L.C.; Lopes, J.R.; de Campos Zuccari, D.A.P. Glutathione and glutathione peroxidase expression in breast cancer: An immunohistochemical and molecular study. Oncol. Rep. 2013, 30, 1119–1128.
- Meng, Q.; Shi, S.; Liang, C.; Liang, D.; Hua, J.; Zhang, B.; Xu, J.; Yu, X. Abrogation of glutathione peroxidase-1 drives EMT and chemoresistance in pancreatic cancer by activating ROS-mediated Akt/GSK3β/Snail signaling. Oncogene 2018, 37, 5843–5857.
- Chang, X.-Z.; Li, D.-Q.; Hou, Y.-F.; Wu, J.; Lu, J.-S.; Di, G.-H.; Jin, W.; Ou, Z.-L.; Shen, Z.-Z.; Shao, Z.-M. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007, 9, 1–15.
- Park, M.H.; Jo, M.; Kim, Y.; Lee, C.; Hong, J. Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 2016, 163, 1–23.
- Lillig, C.H.; Holmgren, A. Thioredoxin and Related Molecules–From Biology to Health and Disease. Antioxid. Redox Signal. 2007, 9, 25–47.
- Nordberg, J.; Arnér, E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system1 1. This review is based on the licentiate thesis “Thioredoxin Reductase—Interactions with the Redox Active Compounds 1-chloro-2,4-dinitrobenzene and lipoic acid” by Jonas Nordberg, 2001, Karolinska Institute, Stockholm, ISBN 91-631-1064-4. Free. Radic. Biol. Med. 2001, 31, 1287–1312.
- Cadenas, C.; Franckenstein, D.; Schmidt, M.; Gehrmann, M.; Hermes, M.; Geppert, B.; Schormann, W.; Maccoux, L.J.; Schug, M.; Schumann, A.; et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 2010, 12, R44.
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free. Radic. Biol. Med. 2014, 66, 75–87.
- Kim, S.J.; Miyoshi, Y.; Taguchi, T.; Tamaki, Y.; Nakamura, H.; Yodoi, J.; Kato, K.; Noguchi, S. High Thioredoxin Expression Is Associated with Resistance to Docetaxel in Primary Breast Cancer. Clin. Cancer Res. 2005, 11, 8425–8430.
- Harris, A.L.; Generali, D. Inhibitors of tumor angiogenesis. Cancer Drug Des. Discov. 2014, 275–317.
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12.
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196.
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352.
- Smith, P.F.; Alberts, D.W.; Rush, G.F. Role of glutathione reductase during menadione-induced NADPH oxidation in isolated rat hepatocytes. Biochem. Pharmacol. 1987, 36, 3879–3884.
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in Cancer Biology and Therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181.
- McIlwain, C.C.; Townsend, D.M.; Tew, K.D. Glutathione S-transferase polymorphisms: Cancer incidence and therapy. Oncogene 2006, 25, 1639–1648.
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375.
- Fujitani, N.; Yoneda, A.; Takahashi, M.; Takasawa, A.; Aoyama, T.; Miyazaki, T. Retracted article: Silencing of Glutathione S-Transferase Pi Inhibits Cancer Cell Growth via Oxidative Stress Induced by Mitochondria Dysfunction. Sci. Rep. 2019, 9, 14764.
- Zhang, K.H.; Tian, H.Y.; Gao, X.; Lei, W.W.; Hu, Y.; Wang, D.M.; Pan, X.C.; Yu, M.L.; Xu, G.J.; Zhao, F.K.; et al. Ferritin heavy chain-mediated iron homeostasis and subsequent increased reactive oxygen species production are essential for epithelial-mesenchymal transition. Cancer Res. 2009, 69, 5340–5348.
- Kiessling, M.K.; Klemke, C.D.; Kamiński, M.M.; Galani, I.E.; Krammer, P.H.; Gülow, K. Inhibition of Constitutively Activated Nuclear Factor-κB Induces Reactive Oxygen Species- and Iron-Dependent Cell Death in Cutaneous T-Cell Lymphoma. Cancer Res. 2009, 69, 2365–2374.
- Shpyleva, S.I.; Tryndyak, V.P.; Kovalchuk, O.; Starlard-Davenport, A.; Chekhun, V.F.; Beland, F.A.; Pogribny, I.P. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res. Treat. 2011, 126, 63–71.



