The idea that Vitamin C (Vit-C) could be utilized as a form of anti-cancer therapy has generated many contradictory arguments. Insights into the physiological characteristics of Vit-C, its pharmacokinetics, and results from preclinical reports, however, suggest that high-dose Vit-C could be effectively utilized in the management of various tumor types. Studies have shown that the pharmacological action of Vit-C can attack various processes that cancerous cells use for their growth and development.
- high-dose
- anti-cancer
- vitamin-C
- pharmacological
- pharmacokinetics
- cancer
- immunotherapy
- ROS-scavenging systems
1. Introduction
2. Historical Background and Justification of High-Dose Vitamin-C in Cancer Management
The notion that Vit-C may play a significant part in cancer prevention was originally postulated in the 1970s by Cameron and colleagues, who suggested that [17,18,19][17][18][19] high-dose Vit-C may increase the survival of cancer patients in their terminal stages. In the same decade, Pauling and Cameron conducted the first recorded research in which Vit-C was given to cancer patients. They demonstrated that giving 100 terminally sick cancer patients with 10 g of Vit-C daily led to excellent results, when compared to a thousand cancer patients who received the conventional treatment. It was shown that 10% of cancer patients who received Vit-C lived; while no malignancy cases that received the conventional treatment without Vit-C went on to survive [18]. The follow-up investigations supported these data. Murata and Morishige demonstrated, in research performed on Japanese patients with uterine malignancies and who received 5 to 30 g of Vit-C daily, that these individuals lived six times longer than those who received only 4 g of Vit-C daily. When a comparison was conducted between individuals who received Vit-C supplements and those who did not, it was shown that those who received Vit-C supplements had a 15% greater survival rate [20]. However, Moertel et al. found that high-dose Vit-C at 10 g per day had no advantage, when compared to placebo in two different investigations on 100 and 150 patients with late stage colorectal cancer (CRC) and patients with late stage cancers, respectively [21,22][21][22]. Now, new findings support the notion that high-dose consumption of Vit-C is associated with a decreased risk of the oral cavity, stomach, esophagus, pancreas, cervix, breast, and rectum cancers [23[23][24],24], and cancers with non-hormonal origins [25]. Furthermore, in terms of the amenable factors of risk for cancer, food counts as one of the most significant. Several independent study panels and committees have come to the conclusion that a high consumption of fruits and vegetables lowers the risk of developing various forms of cancer [26,27][26][27]; and in particular, it was shown that Vit-C intake was inversely linked to fatality rate [28]. In contrast, the consumption of vitamins A, C, and E, according to a research including 34,000 postmenopausal women, did not seem to be associated with a decreased risk for developing breast cancer [27]. In addition, according to some studies, intravenous Vit-C injection has been shown to be beneficial in the therapy of advanced malignancies [29,30][29][30]. Numerous mechanisms have been suggested to support the role of Vit-C in cancer treatment and prevention, including that of increasing the immune system activity; stimulating collagen synthesis; preventing metastasis, by inhibiting certain enzymatic reaction; inhibiting tumor-causing viruses; correcting for the lack of Vit-C, which is normally linked with cancer patients; wound healing in patients with cancer subsequent to surgery; improving chemotherapy sensitivity; decreasing chemotherapy toxicity; and neutralizing certain carcinogens [31]. Various recent experimental investigations have shown that when tumor cells are exposed to high doses of Vit-C, they suffer growth arrest [32,33][32][33]. Recent reports have also shown that Vit-C administration inhibits metastasis, tumor development, and inflammatory-associated cytokine production, while improving tumor inclusion and enhancing chemotherapy [34,35][34][35]. According to some reports, intravenous injection raises Vit-C levels over 70-fold when compared to oral delivery, and the efficacy of the therapy is inversely proportional to the excess amount of Vit-C [29,36][29][36]. For this reason, the ideal method of administration, the dosage, and the length of the treatment are being intensely debated. Recently reported pharmacokinetic data have improved ourthe knowledge about Vit-C transport regulation and given more clues about the therapeutic effectiveness of Vit-C. This has increased the interest in reconsidering the possibility of utilizing Vit-C in the inhibition of cancer development. Despite the fact that the procedures in these reports vary, the majority of the current research on Vit-C and cancer explores the impact of high-dose Vit-C on the formation and development of cancers, as well as the mechanisms of action that govern the anti-tumor impact of Vit-C [37]. In addition, studies have refocused on the consequences and usefulness of high intravenous Vit-C doses in cancer treatment. In vitro, pharmacological doses of Vit-C from 0.3–20 mmol/L preferentially target and kill cancer cells, compared to the usual physiological levels of Vit-C, which are 0.1 mmol/L. This tumor-killing phenomenon is due to the pro-oxidant characteristics of Vit-C, which, at high concentrations, facilitates the formation of hydrogen peroxide (H2O2), which may be an agent responsible for the anti-tumor impact of Vit-C and its use as a pro-drug in cancer therapies [38,39][38][39]. However, determining the exact contribution of Vit-C to clinical results is challenging, because patients are usually receiving several therapeutic regimens at the same time [40]. As a consequence, although the therapeutic benefit of high-dose Vit-C in cancer remission or development has not been unambiguously established, intravenous (i.v.) administration of the vitamin in high doses can enhance quality of life, even in the advanced stages of the disease [40].3. Different Oxidized Forms of Vitamin-C
Given that Vit-C can occur in several oxidative states, interestingly, ascorbic acid is oxidized by ROS or/and free radicals, resulting in the formation of a reactive anion intermediate radical (Asc•−), which is subsequently oxidized to de-hydroascorbic (DHA) acid [41,42][41][42]. DHA, which has a relatively short half-life of just a few minutes [43], is converted to around 1–5% of the Vit-C inside the human cells [44], and it can be transferred into the cell or hydrolyzed irreversibly into 2,3-Diketo-L-gulonic (2,3-DKG) acid (C6H8O7). When 2,3-DKG is broken down into Ethanedioic acid (C2H2O4) and (2R,3S)-2,3,4-Trihydroxybutanoic acid (C4H8O5), the Vit-C level is significantly decreased [45]. DHA is quickly converted to Vit-C within the cell, by interacting with reduced glutathione (GSH) [45,46,47][45][46][47]. NADPH then recycles the oxidized glutathione (glutathione disulfide (GSSG)) and converts it back into GSH [45].4. Enzymatic Activities of Vitamin-C
The biochemical activities of Vit-C may be ascribed to its chemical characteristics, which can donate electrons to a variety of molecules. Physiological ascorbate, which acts as an anti-oxidant at micromolar concentrations, may reduce ROS toxicity [42,48][42][48]. It may also act as a pro-oxidant at low plasma levels, which might be created via the i.v delivery of Vit-C [49]. Surprisingly, Vit-C also serves as a critical co-factor for many enzymes, easily giving electrons to prosthetic metal ions to attain a complete enzymatic action [44]. Overall, these proteins are divided into two categories: mono-oxygenases-containing copper and a-KG-, oxygen-, and iron-dependent dioxygenasesoxygen-, and iron-dependent dioxygenasesoxygen; and iron-dependent dioxygenases (α-KG; identified as 2-oxoglutarate (2OG))-dependent dioxygenases (α-KGDDs). α-KGDDs are iron-containing proteins that use oxygen (O2) as well as α-KG as an enzyme co-factor to produce succinate and CO2. α-KGDDs also catalyze various chemical processes involved in the construction of collagen, hypoxia-inducible factor 1-alpha (HIF1-α) stability, carnitine synthesis, tyrosine catabolism, and protein, DNA, and RNA de-methylation [50,51,52][50][51][52]. Accordingly, Vit-C is crucial for regulating a wide range of essential cellular activities.5. ROS
As the therapeutic impact of high-dose Vit-C is strongly dependent on the formation of ROS, it is essential to define this term. The term “ROS” refers to a group of highly reactive chemical species formed when electrons escape from the mitochondrial electron transport chain (ETC; coenzyme Q) and interact with molecular O2, which is converted enzymatically to superoxide (O2•−) and dismutated to produce H2O2, which is then partially reduced to form hydroxide ions (OH−), hydroxyl radicals (HO•), and water (H2O) [53,54][53][54]. It is worth noting that the superoxide and the hydroxyl radical are together referred to as ROS free radicals [54]. Furthermore, a broad spectrum of substances may produce ROS exogenously. These include heavy metals, radiation, cigarette smoke, drugs, and xenosensors [55,56[55][56][57][58][59],57,58,59], among others. ROS play a key regulatory function in various metabolic pathways in living systems; hence, they are being continuously generated and eliminated. Under normal physiological circumstances, cells control ROS levels through their scavenging machinery [60]. However, under oxidative pressure, increased levels of ROS can result in DNA, protein, and lipid damage, which can potentially lead to cancer formation [61]. However, the action of ROS is essential for eradicating cancers at the molecular level. For these reasons, strategies to increase or eliminate ROS have been established. Tumors with higher ROS levels, for example, depend largely on their anti-oxidative stress defense systems. Drugs that elevate ROS can increase intercellular ROS amounts directly through ROS production (e.g., gadolinium, motexafin, elesclomol) or through inhibition of superoxide dusmutases (SODs) (e.g., ATN-224, 2-methoxyestradiol), and GSH (β-phenylethyl isothiocyanate (PEITC), buthionine sulfoximine (BSO)), which can produce massive ROS amounts, leading to cancer cell death [62,63][62][63]. However, under the low basal stress, normal cells appear to respond in a positive manner to enhanced ROS levels [63]. As a result, increasing ROS levels in all cells may be employed to selectively destroy cancer cells, a strategy discussed here in the context of high-dose Vit-C therapies.6. Anti-Cancer Mechanisms of Vitamin-C
In recent years, numerous experimental reports have confirmed that low, millimolar range doses of pharmacological Vit-C may destroy tumors in vitro and prevent cancer development in vivo. The precise process by which certain tumor cells become susceptible to Vit-C, whereas wild type cells are unaffected is unclear. In fact, there is a wide range of mechanisms through which Vit-C may impact the progression of cancer. Conversely, the efficacy of this interaction is affected by a range of variables, such as the type of malignancy being managed and the different pathways that tumor cells utilize. In this section, wresearchers will look at how the anticancer mechanism of pharmacological ascorbate affects cancer cells. First, cancer cells, owing to their defective mitochondria and increased metabolic reliance, are more sensitive to oxidative stress than normal body cells [64]. Oxidative stress may aid in tumor growth via ROS, by enhancing the cell development and increasing the genetic imbalance. However, an excess of ROS may destroy cancer, and to avoid this deleterious effect, cancer cells utilize a variety of mechanisms that may block the negative effects of ROS [42,65][42][65]. Since cancer cells can utilize ROS for their development and growth [66], anti-oxidant treatments have been investigated as potential anti-cancer strategies, based on the notion that ROS promotes tumor development. Various studies, on the other hand, have shown no convincing indication of the effectiveness of antioxidant therapy in inhibiting tumor growth [67]. On the contrary, the antioxidant treatments seemed to facilitate cancer development and cancer cells metastasis in animal models [68,69][68][69] and increase the possibility for the development of certain cancers in humans [70,71][70][71]. These outcomes indicate that various malignancies depend on anti-oxidants for survival and, as a result, pro-oxidant drugs may be amenable in this case. Undoubtedly, pro-oxidant anti-tumor treatments, such as radiation, have been used for the treatment of patients with cancer [72]. On the other hand, the existing anti-oxidant therapies often cause substantial collateral damage, making it difficult to use them regularly [72]. Therefore, high-dose Vit-C may avoid these issues by targeting multiple tumor hallmarks, while simultaneously demonstrating no toxicity.7. High-Dose Vitamin-C Enhances Cancer Immunotherapy
Numerous tumors resist the immune response by expressing high amounts of checkpoint proteins, such as programmed cell death 1 (PD-1) and cytotoxic T lymphocyte antigen 4 (CTLA-4) [195][73]. Therefore, anti-checkpoint (ICP) medicines targeting PD-1 or programmed cell death ligand 1 (PD-L1) and CTLA-4 have been licensed for the treatment of a variety of cancers. When used as monotherapy regimens, these medicines significantly increase survival rates and are relatively safe [196][74]. However, treatment failure occurs in more than half of individuals treated with these medicines as monotherapies. CTLA-4 and PD-1 combined inhibition has now been suggested to improve patient response rates and survival rates [196][74]. However, 50% of patients were subject to a higher toxicity as a result of the therapy regimen [197][75]. To reduce the toxicity and maximize the efficacy of these combinations, they must be used with drugs that are efficient in activating immune cells, namely cytotoxic T cells (CTLs), natural killer cells (NK cells), and antigen-presenting cells (APCs). In this regard, high-dose Vit-C has been proven to enhance cancer immunotherapies in several in vivo and in vitro studies. Accordingly, a combination of high-dose Vit-C (4 g/kg) with anti-CTLA-4 (200 μg) and anti-PD-1 (250 μg) antibodies resulted in significant tumor impairment and remission via the infiltration and activation of anti-cancer adaptive immunity (CD8 T and CD4 T cells), especially CD8 T cells. This activation was revealed by the production of higher interferon-gamma (IFN-γ) and the increased effectiveness of immune checkpoint inhibitors in various animals and in vitro models [35]. Along the same line, after addition of high-dose Vit-C (1.5 M) with anti-PD1 (200 μg) antibodies in a lymphoma mouse model, high-dose Vit-C therapy improved tumor immune recognition, and resulted in enhanced macrophage and cytotoxic T cell infiltration, which is not seen with anti-PD1 treatment alone. In addition, anti-PD1 antibodies inhibited the PD-1/PD-L1 axis’s inhibitory effect on APCs, CD8+ T, and NK cells. However, anti-PD1 alone is more effective in directly activating these cells than high-dose Vit-C. Indeed, the combined therapy significantly increased the production of IL-12 by APCs and granzyme B by cytotoxic cells (CD8+ T cells and NK cells) as compared to either of the drugs alone [198][76]. An additional study in the RCC mice model demonstrated that high-dose Vit-C therapy alone led to tumor infiltration by both CD4+ and CD8+ T cells and elevated the ratio CD8+/CD4+, while regulating the production of numerous cytokines and chemokines. Anti-PD-L1 antibody injection alone increased intratumoral CD4+ and CD8+ T cells significantly. However, the infiltration of CD4+ and CD8+ T cells, as well as the CD8+/CD4+ ratio, increased considerably when high doses of Vit-C were coupled with anti-PD-L1 antibodies. Specifically, Vit-C stimulates TET2, which in turn activates IRF1, which eventually binds to STAT1, to enhance the expression of PD-L, making tumor cells more susceptible to immunotherapy [199][77]. These findings indicate that high-dose Vit-C and the anti-PD-L1 antibody can act synergistically to eliminate tumor cells, and that the immunotherapy effect of Vit-C-anti-PD-L1 combination is TET2-dependent. Supporting this notion, in mouse melanoma and colon tumors, high-dose Vit-C dramatically enhanced T helper 1 (Th1) chemokines and tumor-infiltrating lymphocytes in the IFN-γ/JAK/STAT/TET axis, resulting in improved anti-tumor immunity and anti-PD-L1 effectiveness. Specifically, Vit-C enhanced IFN-γ-stimulated production of three Th1-type chemokines and PD-L1 genes in a TET2-dependent sense, where the activated TET2 binds to and increases the levels of 5hmC in the CXCL10 and PD-L1 promoters. However, loss of TET2 significantly decreased the ability of Vit-C to activate tumor-infiltrating CD8+ and CD3+ cells, indicating that TET2 is the primary target for Vit-C in enhancing the effectiveness of the anti-PD-L1 therapy [200][78]. TET activity may be used as a biomarker to predict the efficacy and response of patients to an anti-PD-1/PD-L1 therapy, and high-dose Vit-C could be regarded as an adjuvant to immunotherapy through the achieved stimulation of the TET activity, especially for solid tumors expressing significantly lower levels of 5hmC. Based on the ability of high-dose Vit-C to produce massive amounts of ROS in tumor cells, a recent study reported that Vit-C and oncolytic adenoviruse (oAds) combination therapy increased the production of ROS significantly, as compared to Vit-C or oAds monotherapies. This increased production of ROS, in turn, induced immunogenic cell death (ICD), which was confirmed by the increase in the calreticulin (CRT) translocation onto the cell membrane and by the upregulation of two ICD markers, heat shock protein 90 (HSP90) and high mobility group box 1 (HMGB1), which successfully stimulated dendritic cell (DC) maturation [201][79]. The results of the combined treatment were further evaluated in a variety of tumor-bearing mice. As a result of DC (CD11c+MHC-II+, CD80+, and CD86+) maturation and activation, the Vit-C and oAds recipe also expanded the numbers of CD3+, CD4+, and CD8+ T cells, particularly CD8+ T cells (IFN-γ, STAT1, CD137, IL-12p35, Granzyme B, and Perforin), in the tumor tissues and caused the upregulation of genes associated with T-cell motility, for example CCL3, CCL4, CCL5, and CXCL10 [201][79]. In addition, the combination therapy also significantly reduced the proportion of Regulatory T cells (Tregs-FoxP3+CD25+) and increased the ratios of CD4+ T cells to Tregs and CD8+ T cells to Tregs. At the same time, neither Vit-C or oAds single-therapy nor Vit-C and oAds combination therapy diminished the ratio of MDSCs (Gr-1+CD11b+) in cancer tissues. However, single-therapy with Vit-C alone or oAds alone reduced the ratio of Tumor-associated macrophages (TAMs-F4/80+CD11b+) in tumor tissues. While the combined Vit-C and oAds reduced the proportion of TAMs (M2 polarized TAMs (CD206+ F4/80+CD11b+) in tumor tissues. All these events, together, mediated the tumor suppression in tumor-bearing mice and in cell culture treated with Vit-C and oAds combined therapy [201][79] (Figure 1).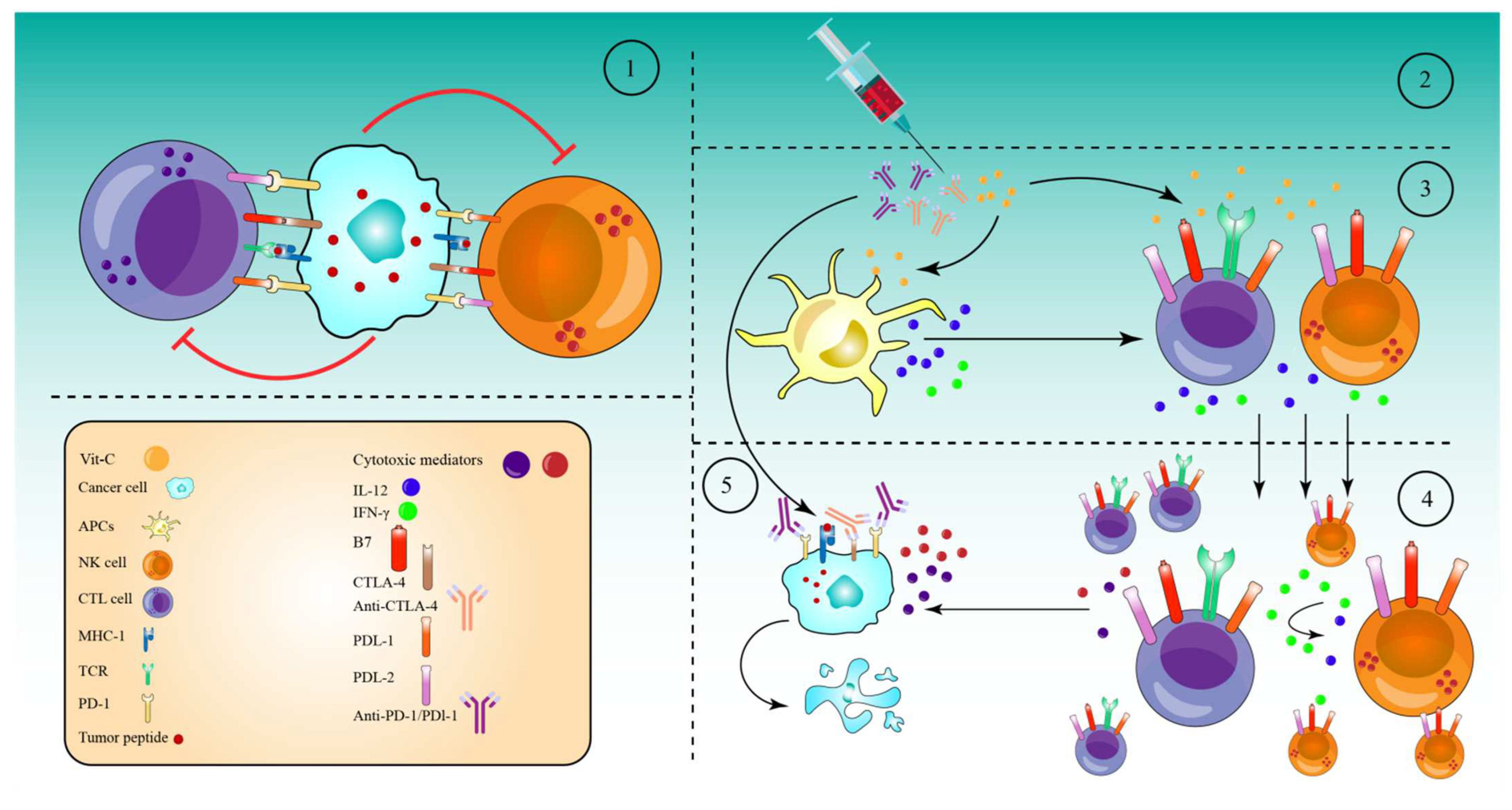
8. Antioxidant Systems That Might Inhibit the Effect of High-Dose Vitamin-C Therapy
Cells use a range of mechanisms to counteract ROS-induced oxidative stress, all of which are mediated by different antioxidant regulatory systems, including CAT, superoxide dismutases (SODs), glutathione peroxidases (GPXs), and Peroxiredoxins (PRDXs) [63,202,203][63][80][81]. Furthermore, these systems convert O2•−, OH•, and H2O2 into H2O and O2 molecules. Additional systems that control ROS include co-factors for the PRDX and GPX-catalyzed processes of reduced GSH and reduced thioredoxin (TRX), respectively; while, GSH is also employed by glutathione-S-transferases (GSTs) to inhibit ROS production (Figure 2), (Table 1) [204][82]. Additionally, cells can regulate the ROS generated by Fenton reaction indirectly through the FTH, which mediates the suppression via LIP sequestration (Figure 2) [205][83], which in turn reduces ROS production. Surprisingly, cancer cells may be able to adapt to the toxicity caused by ROS after receiving a high-dose of ascorbate, by upregulating the intracellular antioxidant proteins and non-enzymatic molecules.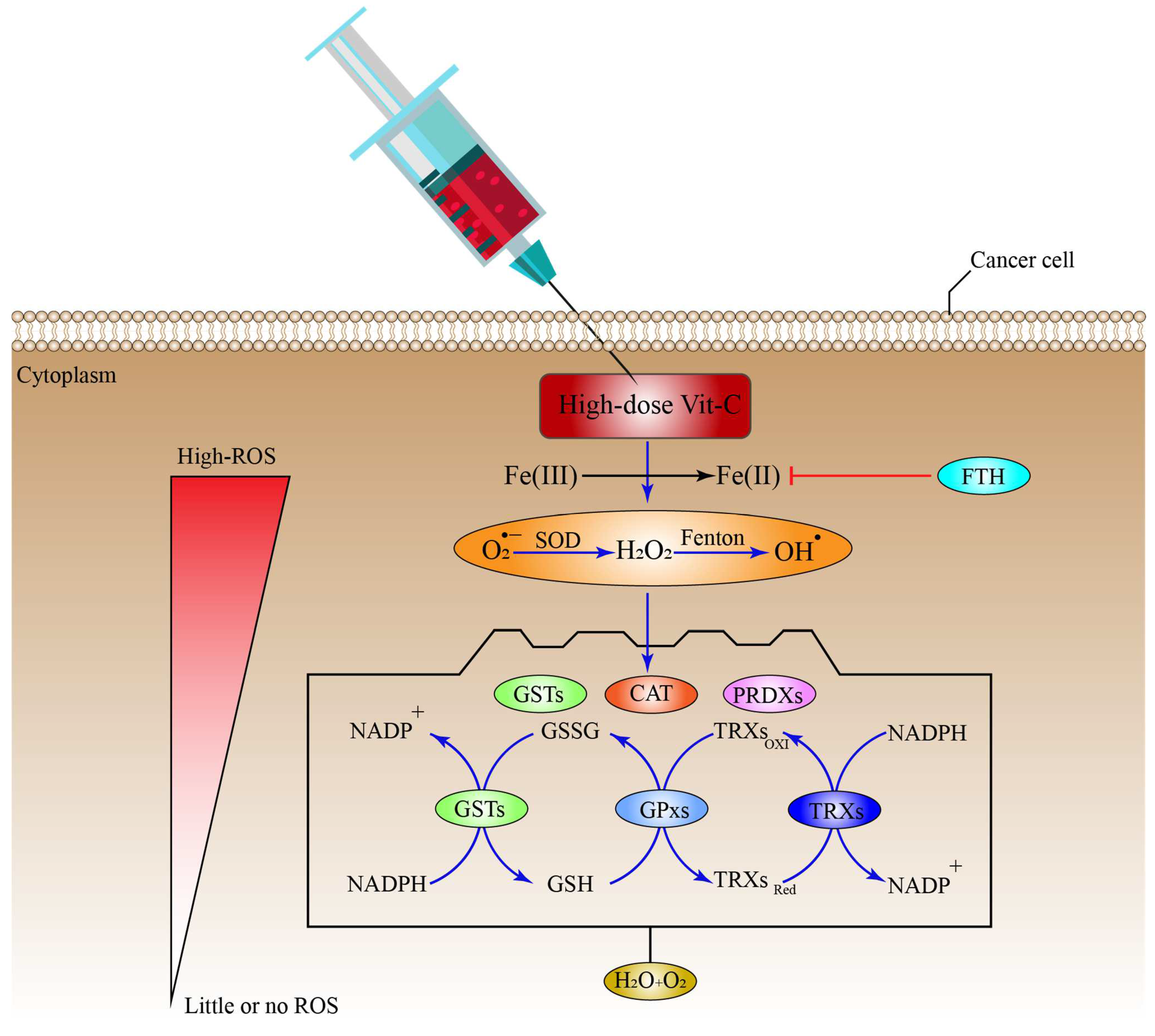
|
Defense System |
Sub-Cellular Localization(s) |
ROS Type and System Function |
Ref. |
|---|---|---|---|
|
SODs |
Cytosol and Peroxisomes |
O2•− to O2 and H2O2 |
|
|
CAT |
Peroxisomes |
H2O2 to H2O and O2 |
|
|
GPxs |
Mitochondria and Cytosol |
H2O2 to H2O lipid peroxides to alcohols |
|
|
PRDXs |
Cytosol, Mitochondria, Nucleus And Endoplasmic reticulum |
H2O2 to H2O |
|
|
TRXs |
Cytosol, Mitochondria, Nucleus |
H2O2 to H2O and O2 |
|
|
GSH |
Cytosol, Mitochondria, Nucleus And Endoplasmic reticulum |
H2O2 to H2O |
|
|
GSTs * |
Cytosol, Membrane-bound |
conjugates with reduced GSH) |
|
|
FTH * |
Nucleus, Lysosome, Cytoplasm |
Sequester Fe(II) to inhibit ROS generation |
SODs, superoxide dismutases; CAT, catalase; PRDXs, Peroxiredoxins; GSTs, glutathione-S-transferases; TRXs, Thioredoxins; GPxs, glutathione peroxidases; GSH, reduced glutathione; GSSG; FTH, ferritin heavy chain, indirect ROS formation inhibition *.
References
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Yagi, K. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-gamma-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 13685–13688.
- Drouin, G.; Godin, J.-R.; Pagé, B. The genetics of vitamin C loss in vertebrates. Curr. Genom. 2011, 12, 371–378.
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396.
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1999, 13, 1007–1024.
- Englard, S.; Seifter, S. The biochemical functions of ascorbic acid. Annu. Rev. Nutr. 1986, 6, 365–406.
- Kuiper, C.; Vissers, M.C.M. Ascorbate as a Co-Factor for Fe- and 2-Oxoglutarate Dependent Dioxygenases: Physiological Activity in Tumor Growth and Progression. Front. Oncol. 2014, 4, 359.
- Young, J.I.; Züchner, S.; Wang, G. Regulation of the Epigenome by Vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564.
- Carr, A.C.; McCall, C. The role of vitamin C in the treatment of pain: New insights. J. Transl. Med. 2017, 15, 77.
- Ting, H.H.; Timimi, F.K.; Boles, K.S.; Creager, S.J.; Ganz, P.; Creager, M.A. Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1996, 97, 22–28.
- Salonen, R.M.; Nyyssönen, K.; Kaikkonen, J.; Porkkala-Sarataho, E.; Voutilainen, S.; Rissanen, T.H.; Tuomainen, T.P.; Valkonen, V.P.; Ristonmaa, U.; Lakka, H.M.; et al. Six-year effect of combined vitamin C and E supplementation on atherosclerotic progression: The Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) Study. Circulation 2003, 107, 947–953.
- Van Straten, M.; Josling, P. Preventing the common cold with a vitamin C supplement: A double-blind, placebo-controlled survey. Adv. Ther. 2002, 19, 151–159.
- Valero, M.P.; Fletcher, A.E.; De Stavola, B.L.; Vioque, J.; Alepuz, V.C. Vitamin C is associated with reduced risk of cataract in a Mediterranean population. J. Nutr. 2002, 132, 1299–1306.
- Ramdas, W.D.; Schouten, J.; Webers, C.A.B. The Effect of Vitamins on Glaucoma: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 359.
- Seddon, J.M.; Ajani, U.A.; Sperduto, R.D.; Hiller, R.; Blair, N.; Burton, T.C.; Farber, M.D.; Gragoudas, E.S.; Haller, J.; Miller, D.T.; et al. Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group. Jama 1994, 272, 1413–1420.
- Chen, G.C.; Lu, D.B.; Pang, Z.; Liu, Q.F. Vitamin C intake, circulating vitamin C and risk of stroke: A meta-analysis of prospective studies. J. Am. Heart Assoc. 2013, 2, e000329.
- Losonczy, K.G.; Harris, T.B.; Havlik, R.J. Vitamin E and vitamin C supplement use and risk of all-cause and coronary heart disease mortality in older persons: The Established Populations for Epidemiologic Studies of the Elderly. Am. J. Clin. Nutr. 1996, 64, 190–196.
- Zhang, J.; Rao, X.; Li, Y.; Zhu, Y.; Liu, F.; Guo, G.; Luo, G.; Meng, Z.; De Backer, D.; Xiang, H.; et al. Pilot trial of high-dose vitamin C in critically ill COVID-19 patients. Ann. Intensive Care 2021, 11, 5.
- Cameron, E.; Campbell, A. The orthomolecular treatment of cancer. II. Clinical trial of high-dose ascorbic acid supplements in advanced human cancer. Chem. Biol. Interact. 1974, 9, 285–315.
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 3685–3689.
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Reevaluation of prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1978, 75, 4538–4542.
- Murata, A.; Morishige, F.; Yamaguchi, H. Prolongation of survival times of terminal cancer patients by administration of large doses of ascorbate. Int. J. Vitam. Nutr. Res. 1982, 23, 103–113.
- Creagan, E.T.; Moertel, C.G.; O’Fallon, J.R.; Schutt, A.J.; O’Connell, M.J.; Rubin, J.; Frytak, S. Failure of High-Dose Vitamin C (Ascorbic Acid) Therapy to Benefit Patients with Advanced Cancer. N. Engl. J. Med. 1979, 301, 687–690.
- Moertel, C.G.; Fleming, T.R.; Creagan, E.T.; Rubin, J.; O’Connell, M.J.; Ames, M.M. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N. Engl. J. Med. 1985, 312, 137–141.
- Block, G. Vitamin C and cancer prevention: The epidemiologic evidence. Am. J. Clin. Nutr. 1991, 53 (Suppl. 1), 270s–282s.
- Block, G. Epidemiologic evidence regarding vitamin C and cancer. Am. J. Clin. Nutr. 1991, 54 (Suppl. 6), 1310s–1314s.
- Head, K.A. Ascorbic acid in the prevention and treatment of cancer. Altern. Med. Rev. A J. Clin. Ther. 1998, 3, 174–186.
- Block, G.; Patterson, B.; Subar, A. Fruit, vegetables, and cancer prevention: A review of the epidemiological evidence. Nutr. Cancer 1992, 18, 1–29.
- Steinmetz, K.A.; Potter, J.D. Vegetables, fruit, and cancer prevention: A review. J. Am. Diet. Assoc. 1996, 96, 1027–1039.
- Loria, C.M.; Klag, M.J.; Caulfield, L.E.; Whelton, P.K. Vitamin C status and mortality in US adults. Am. J. Clin. Nutr. 2000, 72, 139–145.
- Padayatty, S.J.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Hoffer, L.J.; Levine, M. Intravenously administered vitamin C as cancer therapy: Three cases. CMAJ 2006, 174, 937–942.
- Dachs, G.U.; Gandhi, J.; Wohlrab, C.; Carr, A.C.; Morrin, H.R.; Pullar, J.M.; Bayer, S.B.; Eglinton, T.W.; Robinson, B.A.; Vissers, M.C.M. Vitamin C Administration by Intravenous Infusion Increases Tumor Ascorbate Content in Patients With Colon Cancer: A Clinical Intervention Study. Front. Oncol. 2021, 10, 600715.
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609.
- Cabanillas, F. Vitamin C and cancer: What can we conclude—1609 patients and 33 years later? Puerto Rico Health Sci. J. 2010, 29, 215–217.
- Cha, J.; Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Ascorbate depletion increases growth and metastasis of melanoma cells in vitamin C deficient mice. Exp. Oncol. 2011, 33, 226–230.
- Cha, J.; Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Ascorbate supplementation inhibits growth and metastasis of B16FO melanoma and 4T1 breast cancer cells in vitamin C-deficient mice. Int. J. Oncol. 2013, 42, 55–64.
- Magrì, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chilà, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707.
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537.
- Kushi, L.H.; Fee, R.M.; Sellers, T.A.; Zheng, W.; Folsom, A.R. Intake of vitamins A, C, and E and postmenopausal breast cancer. The Iowa Women’s Health Study. Am. J. Epidemiol. 1996, 144, 165–174.
- Roa, F.J.; Pena, E.; Gatica, M.; Escobar-Acuna, K.; Saavedra, P.; Maldonado, M.; Cuevas, M.; Moraga-Cid, G.; Rivas, C.I.; Munoz-Montesino, C. Therapeutic Use of Vitamin C in Cancer: Physiological Considerations. Front. Pharmacol. 2020, 11, 211.
- Assouline, S.; Miller, W.H. High-dose vitamin C therapy: Renewed hope or false promise? CMAJ 2006, 174, 956–957.
- Yeom, C.H.; Jung, G.C.; Song, K.J. Changes of terminal cancer patients’ health-related quality of life after high dose vitamin C administration. J. Korean Med. Sci. 2007, 22, 7–11.
- Corti, A.; Casini, A.F.; Pompella, A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch. Biochem. Biophys. 2010, 500, 107–115.
- Glorieux, C.; Buc Calderon, P. Vitamin C (Ascorbate) and Redox Topics in Cancer. Antioxid Redox Signal 2021, 35, 1157–1175.
- Vera, J.C.; Rivas, C.I.; Zhang, R.H.; Farber, C.M.; Golde, D.W. Human HL-60 myeloid leukemia cells transport dehydroascorbic acid via the glucose transporters and accumulate reduced ascorbic acid. Blood 1994, 84, 1628–1634.
- Padayatty, S.J.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral Dis. 2016, 22, 463–493.
- Wilson, J.X. The physiological role of dehydroascorbic acid. FEBS Lett. 2002, 527, 5–9.
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; Del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965.
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543.
- Podmore, I.D.; Griffiths, H.R.; Herbert, K.E.; Mistry, N.; Mistry, P.; Lunec, J. Vitamin C exhibits pro-oxidant properties. Nature 1998, 392, 559.
- Flashman, E.; Davies, S.L.; Yeoh, K.K.; Schofield, C.J. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem. J. 2010, 427, 135–142.
- Dao, J.H.; Kurzeja, R.J.; Morachis, J.M.; Veith, H.; Lewis, J.; Yu, V.; Tegley, C.M.; Tagari, P. Kinetic characterization and identification of a novel inhibitor of hypoxia-inducible factor prolyl hydroxylase 2 using a time-resolved fluorescence resonance energy transfer-based assay technology. Anal. Biochem. 2009, 384, 213–223.
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free. Radic. Biol. Med. 2015, 82, 22–28.
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824.
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175.
- Muthukumar, K.; Nachiappan, V. Cadmium-induced oxidative stress in Saccharomyces cerevisiae. Indian J. Biochem. Biophys. 2010, 47, 383–387.
- Tominaga, H.; Kodama, S.; Matsuda, N.; Suzuki, K.; Watanabe, M. Involvement of reactive oxygen species (ROS) in the induction of genetic instability by radiation. J. Radiat. Res. 2004, 45, 181–188.
- van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.L.; Gans, R.O.B.; Koëter, G.H.; Choi, A.M.K.; van Oosterhout, A.J.M.; Slebos, D.-J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung. Cell Mol. Physiol. 2009, 297, L109–L114.
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-Induced Oxidative Stress and Toxicity. J. Toxicol. 2012, 2012, 645460.
- Klotz, L.-O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654.
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511.
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free. Radic Res. 2010, 44, 479–496.
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176.
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. CB 2014, 24, R453–R462.
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429.
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735.
- Thyagarajan, A.; Sahu, R.P. Potential Contributions of Antioxidants to Cancer Therapy: Immunomodulation and Radiosensitization. Integr. Cancer Ther. 2018, 17, 210–216.
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra215.
- Zou, Z.V.; Le Gal, K.; El Zowalaty, A.E.; Pehlivanoglu, L.E.; Garellick, V.; Gul, N.; Ibrahim, M.X.; Bergh, P.-O.; Henricsson, M.; Wiel, C.; et al. Antioxidants Promote Intestinal Tumor Progression in Mice. Antioxidants 2021, 10, 241.
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556.
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L.; Valanis, B.; Williams, J.H.; et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N. Engl. J. Med. 1996, 334, 1150–1155.
- Wondrak, G.T. Redox-directed cancer therapeutics: Molecular mechanisms and opportunities. Antioxid Redox Signal 2009, 11, 3013–3069.
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11.
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255.
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16.
- Luchtel, R.A.; Bhagat, T.; Pradhan, K.; Jacobs, W.R.; Levine, M.; Verma, A.; Shenoy, N. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl. Acad. Sci. USA 2020, 117, 1666–1677.
- Peng, D.; He, A.; He, S.; Ge, G.; Wang, S.; Ci, W.; Li, X.; Xia, D.; Zhou, L. Ascorbic acid induced TET2 enzyme activation enhances cancer immunotherapy efficacy in renal cell carcinoma. Int. J. Biol. Sci. 2022, 18, 995–1007.
- Xu, Y.-p.; Lv, L.; Liu, Y.; Smith, M.D.; Li, W.-C.; Tan, X.-m.; Cheng, M.; Li, Z.; Bovino, M.; Aubé, J.; et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Investig. 2019, 129, 4316–4331.
- Ma, J.; Zhang, C.; Shi, G.; Yue, D.; Shu, Y.; Hu, S.; Qi, Z.; Chen, Y.; Zhang, B.; Zhang, Y.; et al. High-dose VitC plus oncolytic adenoviruses enhance immunogenic tumor cell death and reprogram tumor immune microenvironment. Mol. Ther. 2022, 30, 644–661.
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293.
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153.
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297.
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin Heavy Chain Upregulation by NF-κB Inhibits TNFα-Induced Apoptosis by Suppressing Reactive Oxygen Species. Cell 2004, 119, 529–542.
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93.
- Ekoue, D.N.; He, C.; Diamond, A.M.; Bonini, M.G. Manganese superoxide dismutase and glutathione peroxidase-1 contribute to the rise and fall of mitochondrial reactive oxygen species which drive oncogenesis. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1858, 628–632.
- Dhar, S.K.; St. Clair, D.K. Manganese superoxide dismutase regulation and cancer. Free. Radic. Biol. Med. 2012, 52, 2209–2222.
- Becuwe, P.; Ennen, M.; Klotz, R.; Barbieux, C.; Grandemange, S. Manganese superoxide dismutase in breast cancer: From molecular mechanisms of gene regulation to biological and clinical significance. Free. Radic. Biol. Med. 2014, 77, 139–151.
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090.
- Bauer, G. Tumor Cell-protective Catalase as a Novel Target for Rational Therapeutic Approaches Based on Specific Intercellular ROS Signaling. Anticancer. Res. 2012, 32, 2599–2624.
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108.
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997.
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3289–3303.
- Liu, J.; Hinkhouse, M.M.; Sun, W.; Weydert, C.J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. GoswamiRedox Regulation of Pancreatic Cancer Cell Growth: Role of Glutathione Peroxidase in the Suppression of the Malignant Phenotype. Hum. Gene Ther. 2004, 15, 239–250.
- Jardim, B.V.; Moschetta, M.G.; Leonel, C.; Gelaleti, G.B.; Regiani, V.R.; Ferreira, L.C.; Lopes, J.R.; de Campos Zuccari, D.A.P. Glutathione and glutathione peroxidase expression in breast cancer: An immunohistochemical and molecular study. Oncol. Rep. 2013, 30, 1119–1128.
- Meng, Q.; Shi, S.; Liang, C.; Liang, D.; Hua, J.; Zhang, B.; Xu, J.; Yu, X. Abrogation of glutathione peroxidase-1 drives EMT and chemoresistance in pancreatic cancer by activating ROS-mediated Akt/GSK3β/Snail signaling. Oncogene 2018, 37, 5843–5857.
- Chang, X.-Z.; Li, D.-Q.; Hou, Y.-F.; Wu, J.; Lu, J.-S.; Di, G.-H.; Jin, W.; Ou, Z.-L.; Shen, Z.-Z.; Shao, Z.-M. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007, 9, 1–15.
- Park, M.H.; Jo, M.; Kim, Y.; Lee, C.; Hong, J. Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 2016, 163, 1–23.
- Lillig, C.H.; Holmgren, A. Thioredoxin and Related Molecules–From Biology to Health and Disease. Antioxid. Redox Signal. 2007, 9, 25–47.
- Nordberg, J.; Arnér, E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system1 1. This review is based on the licentiate thesis “Thioredoxin Reductase—Interactions with the Redox Active Compounds 1-chloro-2,4-dinitrobenzene and lipoic acid” by Jonas Nordberg, 2001, Karolinska Institute, Stockholm, ISBN 91-631-1064-4. Free. Radic. Biol. Med. 2001, 31, 1287–1312.
- Cadenas, C.; Franckenstein, D.; Schmidt, M.; Gehrmann, M.; Hermes, M.; Geppert, B.; Schormann, W.; Maccoux, L.J.; Schug, M.; Schumann, A.; et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 2010, 12, R44.
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free. Radic. Biol. Med. 2014, 66, 75–87.
- Kim, S.J.; Miyoshi, Y.; Taguchi, T.; Tamaki, Y.; Nakamura, H.; Yodoi, J.; Kato, K.; Noguchi, S. High Thioredoxin Expression Is Associated with Resistance to Docetaxel in Primary Breast Cancer. Clin. Cancer Res. 2005, 11, 8425–8430.
- Harris, A.L.; Generali, D. Inhibitors of tumor angiogenesis. Cancer Drug Des. Discov. 2014, 275–317.
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12.
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196.
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352.
- Smith, P.F.; Alberts, D.W.; Rush, G.F. Role of glutathione reductase during menadione-induced NADPH oxidation in isolated rat hepatocytes. Biochem. Pharmacol. 1987, 36, 3879–3884.
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in Cancer Biology and Therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181.
- McIlwain, C.C.; Townsend, D.M.; Tew, K.D. Glutathione S-transferase polymorphisms: Cancer incidence and therapy. Oncogene 2006, 25, 1639–1648.
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375.
- Fujitani, N.; Yoneda, A.; Takahashi, M.; Takasawa, A.; Aoyama, T.; Miyazaki, T. Retracted article: Silencing of Glutathione S-Transferase Pi Inhibits Cancer Cell Growth via Oxidative Stress Induced by Mitochondria Dysfunction. Sci. Rep. 2019, 9, 14764.
- Zhang, K.H.; Tian, H.Y.; Gao, X.; Lei, W.W.; Hu, Y.; Wang, D.M.; Pan, X.C.; Yu, M.L.; Xu, G.J.; Zhao, F.K.; et al. Ferritin heavy chain-mediated iron homeostasis and subsequent increased reactive oxygen species production are essential for epithelial-mesenchymal transition. Cancer Res. 2009, 69, 5340–5348.
- Kiessling, M.K.; Klemke, C.D.; Kamiński, M.M.; Galani, I.E.; Krammer, P.H.; Gülow, K. Inhibition of Constitutively Activated Nuclear Factor-κB Induces Reactive Oxygen Species- and Iron-Dependent Cell Death in Cutaneous T-Cell Lymphoma. Cancer Res. 2009, 69, 2365–2374.
- Shpyleva, S.I.; Tryndyak, V.P.; Kovalchuk, O.; Starlard-Davenport, A.; Chekhun, V.F.; Beland, F.A.; Pogribny, I.P. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res. Treat. 2011, 126, 63–71.