 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yoon-Seok Roh | -- | 4423 | 2023-11-13 01:35:33 | | | |
| 2 | Jason Zhu | Meta information modification | 4423 | 2023-11-13 06:56:42 | | |
Video Upload Options
Chronic liver disease (CLD) affects a significant portion of the global population, leading to a substantial number of deaths each year. Distinct forms like non-alcoholic fatty liver disease (NAFLD) and alcoholic fatty liver disease (ALD), though they have different etiologies, highlight shared pathologies rooted in oxidative stress. Central to liver metabolism, mitochondria are essential for ATP production, gluconeogenesis, fatty acid oxidation, and heme synthesis. However, in diseases like NAFLD, ALD, and liver fibrosis, mitochondrial function is compromised by inflammatory cytokines, hepatotoxins, and metabolic irregularities. This dysfunction, especially electron leakage, exacerbates the production of reactive oxygen species (ROS), augmenting liver damage. Amidst this, nuclear factor erythroid 2-related factor 2 (NRF2) emerges as a cellular protector. It not only counters oxidative stress by regulating antioxidant genes but also maintains mitochondrial health by overseeing autophagy and biogenesis.
1. Introduction
2. Oxidative Stress and NRF2 Signaling Pathways in Chronic Liver Disease
2.1. The NRF2-KEAP1 Pathway as a Key Regulator of Oxidative Stress in Liver Health
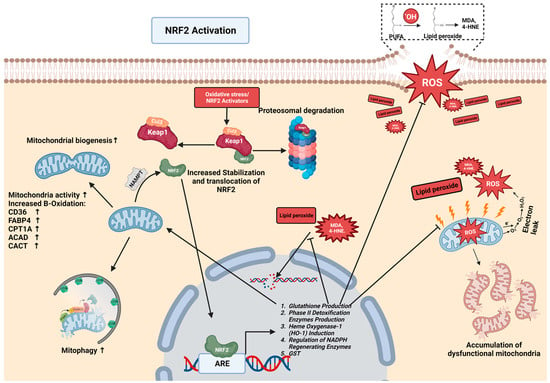
2.2. The NRF2-Mediated Regulation of Lipid Metabolism in CLDs
2.3. NRF2-Mediated Protection against Lipid Peroxidation
2.4. The Interplay of NRF2 and NF-κB in the Modulation of Inflammation via Kupffer Cells
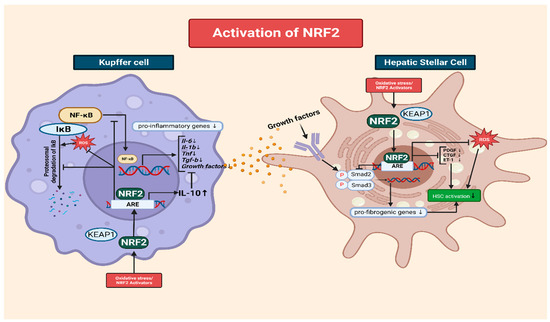
2.5. The Role of NRF2 in Mitigating Oxidative-Stress-Induced HSC Activation
3. Crosstalk between NRF2 and Mitochondria Quality Control in Chronic Liver Disease
3.1. The Role of Mitochondria in the Formation of ROS
3.2. The Importance of Mitochondrial Metabolism in the Progression of Chronic Diseases
3.3. NRF2 Mediates Mitophagy and Mitochondrial Turnover
3.4. The Interplay of Mitochondria and NRF2 in CLD
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171.
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647.
- Seen, S. Chronic liver disease and oxidative stress—A narrative review. Expert. Rev. Gastroenterol. Hepatol. 2021, 15, 1021–1035.
- Moon, A.M.; Singal, A.G.; Tapper, E.B. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin. Gastroenterol. Hepatol. 2020, 18, 2650–2666.
- Juanola, O.; Martinez-Lopez, S.; Frances, R.; Gomez-Hurtado, I. Non-Alcoholic Fatty Liver Disease: Metabolic, Genetic, Epigenetic and Environmental Risk Factors. Int. J. Environ. Res. Public Health 2021, 18, 5227.
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224.
- Paik, J.M.; Kabbara, K.; Eberly, K.E.; Younossi, Y.; Henry, L.; Younossi, Z.M. Global burden of NAFLD and chronic liver disease among adolescents and young adults. Hepatology 2022, 75, 1204–1217.
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13.
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441.
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156.
- Wang, X.; Rao, H.; Liu, F.; Wei, L.; Li, H.; Wu, C. Recent Advances in Adipose Tissue Dysfunction and Its Role in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 3300.
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636.
- Buchanan, R.; Sinclair, J.M.A. Alcohol use disorder and the liver. Addiction 2021, 116, 1270–1278.
- Hyun, J.; Han, J.; Lee, C.; Yoon, M.; Jung, Y. Pathophysiological Aspects of Alcohol Metabolism in the Liver. Int. J. Mol. Sci. 2021, 22, 5717.
- Tan, H.K.; Yates, E.; Lilly, K.; Dhanda, A.D. Oxidative stress in alcohol-related liver disease. World J. Hepatol. 2020, 12, 332–349.
- Kruman, I.I.; Henderson, G.I.; Bergeson, S.E. DNA damage and neurotoxicity of chronic alcohol abuse. Exp. Biol. Med. 2012, 237, 740–747.
- Smathers, R.L.; Galligan, J.J.; Stewart, B.J.; Petersen, D.R. Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. Chem. Biol. Interact. 2011, 192, 107–112.
- Malnick, S.D.H.; Alin, P.; Somin, M.; Neuman, M.G. Fatty Liver Disease-Alcoholic and Non-Alcoholic: Similar but Different. Int. J. Mol. Sci. 2022, 23, 16226.
- Mallet, M.; Silaghi, C.A.; Sultanik, P.; Conti, F.; Rudler, M.; Ratziu, V.; Thabut, D.; Pais, R. Current challenges and future perspectives in treating patients with NAFLD-related cirrhosis. Hepatology 2023.
- Gines, P.; Krag, A.; Abraldes, J.G.; Sola, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376.
- Louvet, A.; Mathurin, P. Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242.
- Ramakrishna, G.; Rastogi, A.; Trehanpati, N.; Sen, B.; Khosla, R.; Sarin, S.K. From cirrhosis to hepatocellular carcinoma: New molecular insights on inflammation and cellular senescence. Liver Cancer 2013, 2, 367–383.
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345.
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777.
- Galicia-Moreno, M.; Lucano-Landeros, S.; Monroy-Ramirez, H.C.; Silva-Gomez, J.; Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Roles of Nrf2 in Liver Diseases: Molecular, Pharmacological, and Epigenetic Aspects. Antioxidants 2020, 9, 980.
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20.
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell. Res. 2018, 1865, 721–733.
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182.
- Shaw, P.; Chattopadhyay, A. Nrf2-ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell. Physiol. 2020, 235, 3119–3130.
- Zhou, J.; Zheng, Q.; Chen, Z. The Nrf2 Pathway in Liver Diseases. Front. Cell. Dev. Biol. 2022, 10, 826204.
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144.
- Qiu, S.; Liang, Z.; Wu, Q.; Wang, M.; Yang, M.; Chen, C.; Zheng, H.; Zhu, Z.; Li, L.; Yang, G. Hepatic lipid accumulation induced by a high-fat diet is regulated by Nrf2 through multiple pathways. FASEB J. 2022, 36, e22280.
- Akl, M.G.; Li, L.; Baccetto, R.; Phanse, S.; Zhang, Q.; Trites, M.J.; McDonald, S.; Aoki, H.; Babu, M.; Widenmaier, S.B. Complementary gene regulation by NRF1 and NRF2 protects against hepatic cholesterol overload. Cell Rep. 2023, 42, 112399.
- Zhang, Y.K.; Yeager, R.L.; Tanaka, Y.; Klaassen, C.D. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharmacol. 2010, 245, 326–334.
- Tanaka, Y.; Ikeda, T.; Yamamoto, K.; Ogawa, H.; Kamisako, T. Dysregulated expression of fatty acid oxidation enzymes and iron-regulatory genes in livers of Nrf2-null mice. J. Gastroenterol. Hepatol. 2012, 27, 1711–1717.
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188.
- Chambel, S.S.; Santos-Goncalves, A.; Duarte, T.L. The Dual Role of Nrf2 in Nonalcoholic Fatty Liver Disease: Regulation of Antioxidant Defenses and Hepatic Lipid Metabolism. Biomed. Res. Int. 2015, 2015, 597134.
- Morita, M.; Ishida, N.; Uchiyama, K.; Yamaguchi, K.; Itoh, Y.; Shichiri, M.; Yoshida, Y.; Hagihara, Y.; Naito, Y.; Yoshikawa, T.; et al. Fatty liver induced by free radicals and lipid peroxidation. Free Radic. Res. 2012, 46, 758–765.
- Martin-Fernandez, M.; Arroyo, V.; Carnicero, C.; Siguenza, R.; Busta, R.; Mora, N.; Antolin, B.; Tamayo, E.; Aspichueta, P.; Carnicero-Frutos, I.; et al. Role of Oxidative Stress and Lipid Peroxidation in the Pathophysiology of NAFLD. Antioxidants 2022, 11, 2217.
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843.
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972.
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438.
- Winczura, A.; Zdzalik, D.; Tudek, B. Damage of DNA and proteins by major lipid peroxidation products in genome stability. Free Radic. Res. 2012, 46, 442–459.
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855.
- Spiteller, G. Is lipid peroxidation of polyunsaturated acids the only source of free radicals that induce aging and age-related diseases? Rejuvenation Res. 2010, 13, 91–103.
- Negre-Salvayre, A.; Auge, N.; Ayala, V.; Basaga, H.; Boada, J.; Brenke, R.; Chapple, S.; Cohen, G.; Feher, J.; Grune, T.; et al. Pathological aspects of lipid peroxidation. Free Radic. Res. 2010, 44, 1125–1171.
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107.
- Busch, C.J.; Hendrikx, T.; Weismann, D.; Jackel, S.; Walenbergh, S.M.; Rendeiro, A.F.; Weisser, J.; Puhm, F.; Hladik, A.; Goderle, L.; et al. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology 2017, 65, 1181–1195.
- Yadav, U.C.; Ramana, K.V. Regulation of NF-κB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid. Med. Cell. Longev. 2013, 2013, 690545.
- Ma, F.; Luo, S.; Lu, C.; Jiang, X.; Chen, K.; Deng, J.; Ma, S.; Li, Z. The role of Nrf2 in periodontal disease by regulating lipid peroxidation, inflammation and apoptosis. Front. Endocrinol. 2022, 13, 963451.
- Gruber, F.; Ornelas, C.M.; Karner, S.; Narzt, M.S.; Nagelreiter, I.M.; Gschwandtner, M.; Bochkov, V.; Tschachler, E. Nrf2 deficiency causes lipid oxidation, inflammation, and matrix-protease expression in DHA-supplemented and UVA-irradiated skin fibroblasts. Free Radic. Biol. Med. 2015, 88, 439–451.
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64.
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 2019, 65, 37–55.
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218.
- Hao, W.; Li, M.; Cai, Q.; Wu, S.; Li, X.; He, Q.; Hu, Y. Roles of NRF2 in Fibrotic Diseases: From Mechanisms to Therapeutic Approaches. Front. Physiol. 2022, 13, 889792.
- Gong, Y.; Yang, Y. Activation of Nrf2/AREs-mediated antioxidant signalling, and suppression of profibrotic TGF-beta1/Smad3 pathway: A promising therapeutic strategy for hepatic fibrosis—A review. Life Sci. 2020, 256, 117909.
- Yang, H.; Luo, F.; Wei, Y.; Jiao, Y.; Qian, J.; Chen, S.; Gong, Y.; Tang, L. TGR5 protects against cholestatic liver disease via suppressing the NF-κB pathway and activating the Nrf2/HO-1 pathway. Ann. Transl. Med. 2021, 9, 1158.
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995.
- Xue, R.; Qiu, J.; Wei, S.; Liu, M.; Wang, Q.; Wang, P.; Sha, B.; Wang, H.; Shi, Y.; Zhou, J.; et al. Lycopene alleviates hepatic ischemia reperfusion injury via the Nrf2/HO-1 pathway mediated NLRP3 inflammasome inhibition in Kupffer cells. Ann. Transl. Med. 2021, 9, 631.
- Liu, X.; Wang, T.; Liu, X.; Cai, L.; Qi, J.; Zhang, P.; Li, Y. Biochanin A protects lipopolysaccharide/D-galactosamine-induced acute liver injury in mice by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Int. Immunopharmacol. 2016, 38, 324–331.
- Rushworth, S.A.; Chen, X.L.; Mackman, N.; Ogborne, R.M.; O’Connell, M.A. Lipopolysaccharide-induced heme oxygenase-1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J. Immunol. 2005, 175, 4408–4415.
- Liang, W.; Greven, J.; Qin, K.; Fragoulis, A.; Horst, K.; Blasius, F.; Wruck, C.; Pufe, T.; Kobbe, P.; Hildebrand, F.; et al. Sulforaphane Exerts Beneficial Immunomodulatory Effects on Liver Tissue via a Nrf2 Pathway-Related Mechanism in a Murine Model of Hemorrhagic Shock and Resuscitation. Front. Immunol. 2022, 13, 822895.
- Sierra-Filardi, E.; Vega, M.A.; Sanchez-Mateos, P.; Corbi, A.L.; Puig-Kroger, A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 2010, 215, 788–795.
- Wang, L.; He, C. Nrf2-mediated anti-inflammatory polarization of macrophages as therapeutic targets for osteoarthritis. Front. Immunol. 2022, 13, 967193.
- Chen, H.; Qin, J.; Shi, H.; Li, Q.; Zhou, S.; Chen, L. Rhoifolin ameliorates osteoarthritis via the Nrf2/NF-κB axis: In Vitro and in vivo experiments. Osteoarthr. Cartil. 2022, 30, 735–745.
- Liu, G.H.; Qu, J.; Shen, X. NF-κB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727.
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and Their Concerted Modulation in Cancer Pathogenesis and Progression. Cancers 2010, 2, 483–497.
- Nair, S.; Doh, S.T.; Chan, J.Y.; Kong, A.N.; Cai, L. Regulatory potential for concerted modulation of Nrf2- and Nfkb1-mediated gene expression in inflammation and carcinogenesis. Br. J. Cancer 2008, 99, 2070–2082.
- Jiang, T.; Tian, F.; Zheng, H.; Whitman, S.A.; Lin, Y.; Zhang, Z.; Zhang, N.; Zhang, D.D. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-κB-mediated inflammatory response. Kidney Int. 2014, 85, 333–343.
- Heiss, E.; Herhaus, C.; Klimo, K.; Bartsch, H.; Gerhauser, C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J. Biol. Chem. 2001, 276, 32008–32015.
- Kim, J.E.; You, D.J.; Lee, C.; Ahn, C.; Seong, J.Y.; Hwang, J.I. Suppression of NF-κB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation. Cell. Signal. 2010, 22, 1645–1654.
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 367–398.
- Shi, Y.S.; Li, X.X.; Li, H.T.; Zhang, Y. Pelargonidin ameliorates CCl(4)-induced liver fibrosis by suppressing the ROS-NLRP3-IL-1beta axis via activating the Nrf2 pathway. Food Funct. 2020, 11, 5156–5165.
- Prestigiacomo, V.; Suter-Dick, L. Nrf2 protects stellate cells from Smad-dependent cell activation. PLoS ONE 2018, 13, e0201044.
- Qiu, L.; Hu, L.; Liu, X.; Li, W.; Zhang, X.; Xia, H.; Zhang, C. Physalin B inhibits PDGF-BB-induced VSMC proliferation, migration and phenotypic transformation by activating the Nrf2 pathway. Food Funct. 2021, 12, 10950–10966.
- Del Campo, J.A.; Gallego, P.; Grande, L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J. Hepatol. 2018, 10, 1–7.
- Eisenstein, A.; Hilliard, B.K.; Pope, S.D.; Zhang, C.; Taskar, P.; Waizman, D.A.; Israni-Winger, K.; Tian, H.; Luan, H.H.; Wang, A. Activation of the transcription factor NRF2 mediates the anti-inflammatory properties of a subset of over-the-counter and prescription NSAIDs. Immunity 2022, 55, 1082–1095.e1085.
- Kong, D.; Zhang, Z.; Chen, L.; Huang, W.; Zhang, F.; Wang, L.; Wang, Y.; Cao, P.; Zheng, S. Curcumin blunts epithelial-mesenchymal transition of hepatocytes to alleviate hepatic fibrosis through regulating oxidative stress and autophagy. Redox Biol. 2020, 36, 101600.
- Yang, W.; Wang, Y.; Zhang, C.; Huang, Y.; Yu, J.; Shi, L.; Zhang, P.; Yin, Y.; Li, R.; Tao, K. Maresin1 Protect Against Ferroptosis-Induced Liver Injury Through ROS Inhibition and Nrf2/HO-1/GPX4 Activation. Front. Pharmacol. 2022, 13, 865689.
- Silva-Llanes, I.; Shin, C.H.; Jimenez-Villegas, J.; Gorospe, M.; Lastres-Becker, I. The Transcription Factor NRF2 Has Epigenetic Regulatory Functions Modulating HDACs, DNMTs, and miRNA Biogenesis. Antioxidants 2023, 12, 641.
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343.
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15.
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950.
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between mitochondrial membrane potential and ROS formation. Methods Mol. Biol. 2012, 810, 183–205.
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077.
- Xiang, L.; Shao, Y.; Chen, Y. Mitochondrial dysfunction and mitochondrion-targeted therapeutics in liver diseases. J. Drug Target. 2021, 29, 1080–1093.
- Zhang, C.; Zhao, Y.; Yu, M.; Qin, J.; Ye, B.; Wang, Q. Mitochondrial Dysfunction and Chronic Liver Disease. Curr. Issues Mol. Biol. 2022, 44, 3156–3165.
- Koliaki, C.; Roden, M. Hepatic energy metabolism in human diabetes mellitus, obesity and non-alcoholic fatty liver disease. Mol. Cell. Endocrinol. 2013, 379, 35–42.
- Passarella, S.; Schurr, A.; Portincasa, P. Mitochondrial Transport in Glycolysis and Gluconeogenesis: Achievements and Perspectives. Int. J. Mol. Sci. 2021, 22, 12620.
- Stark, R.; Guebre-Egziabher, F.; Zhao, X.; Feriod, C.; Dong, J.; Alves, T.C.; Ioja, S.; Pongratz, R.L.; Bhanot, S.; Roden, M.; et al. A role for mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) in the regulation of hepatic gluconeogenesis. J. Biol. Chem. 2014, 289, 7257–7263.
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810.
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology 2022, 76, 1452–1465.
- Guerra, I.M.S.; Ferreira, H.B.; Melo, T.; Rocha, H.; Moreira, S.; Diogo, L.; Domingues, M.R.; Moreira, A.S.P. Mitochondrial Fatty Acid beta-Oxidation Disorders: From Disease to Lipidomic Studies-A Critical Review. Int. J. Mol. Sci. 2022, 23, 13933.
- Barbier-Torres, L.; Fortner, K.A.; Iruzubieta, P.; Delgado, T.C.; Giddings, E.; Chen, Y.; Champagne, D.; Fernandez-Ramos, D.; Mestre, D.; Gomez-Santos, B.; et al. Silencing hepatic MCJ attenuates non-alcoholic fatty liver disease (NAFLD) by increasing mitochondrial fatty acid oxidation. Nat. Commun. 2020, 11, 3360.
- Fang, E.F.; Waltz, T.B.; Kassahun, H.; Lu, Q.; Kerr, J.S.; Morevati, M.; Fivenson, E.M.; Wollman, B.N.; Marosi, K.; Wilson, M.A.; et al. Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway. Sci. Rep. 2017, 7, 46208.
- Li, Y.; Feng, Y.F.; Liu, X.T.; Li, Y.C.; Zhu, H.M.; Sun, M.R.; Li, P.; Liu, B.; Yang, H. Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol. 2021, 38, 101771.
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid. Redox Signal. 2021, 34, 517–530.
- Mao, Y.; Du, J.; Chen, X.; Al Mamun, A.; Cao, L.; Yang, Y.; Mubwandarikwa, J.; Zaeem, M.; Zhang, W.; Chen, Y.; et al. Maltol Promotes Mitophagy and Inhibits Oxidative Stress via the Nrf2/PINK1/Parkin Pathway after Spinal Cord Injury. Oxid. Med. Cell. Longev. 2022, 2022, 1337630.
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311.
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435.
- Hayashi, G.; Jasoliya, M.; Sahdeo, S.; Sacca, F.; Pane, C.; Filla, A.; Marsili, A.; Puorro, G.; Lanzillo, R.; Brescia Morra, V.; et al. Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum. Mol. Genet. 2017, 26, 2864–2873.
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188.
- Grattagliano, I.; Montezinho, L.P.; Oliveira, P.J.; Fruhbeck, G.; Gomez-Ambrosi, J.; Montecucco, F.; Carbone, F.; Wieckowski, M.R.; Wang, D.Q.; Portincasa, P. Targeting mitochondria to oppose the progression of nonalcoholic fatty liver disease. Biochem. Pharmacol. 2019, 160, 34–45.
- Serviddio, G.; Bellanti, F.; Sastre, J.; Vendemiale, G.; Altomare, E. Targeting mitochondria: A new promising approach for the treatment of liver diseases. Curr. Med. Chem. 2010, 17, 2325–2337.
- Nassir, F.; Ibdah, J.A. Role of mitochondria in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 2136–2142.
- Yu, A.; Zhou, R.; Xia, B.; Dang, W.; Yang, Z.; Chen, X. NAMPT maintains mitochondria content via NRF2-PPARalpha/AMPKalpha pathway to promote cell survival under oxidative stress. Cell. Signal. 2020, 66, 109496.
- Xie, W.; Zhu, T.; Zhou, P.; Xu, H.; Meng, X.; Ding, T.; Nan, F.; Sun, G.; Sun, X. Notoginseng leaf triterpenes ameliorates mitochondrial oxidative injury via the NAMPT-SIRT1/2/3 signaling pathways in cerebral ischemic model rats. J. Ginseng Res. 2023, 47, 199–209.
- Gabande-Rodriguez, E.; Gomez de Las Heras, M.M.; Mittelbrunn, M. Control of Inflammation by Calorie Restriction Mimetics: On the Crossroad of Autophagy and Mitochondria. Cells 2019, 9, 82.
- Wang, S.; Wan, T.; Ye, M.; Qiu, Y.; Pei, L.; Jiang, R.; Pang, N.; Huang, Y.; Liang, B.; Ling, W.; et al. Nicotinamide riboside attenuates alcohol induced liver injuries via activation of SirT1/PGC-1alpha/mitochondrial biosynthesis pathway. Redox Biol. 2018, 17, 89–98.
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33.

