Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Filippos Triposkiadis | -- | 5179 | 2023-11-08 23:45:09 | | | |
| 2 | Camila Xu | Meta information modification | 5179 | 2023-11-09 02:20:38 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Triposkiadis, F.; Sarafidis, P.; Briasoulis, A.; Magouliotis, D.E.; Athanasiou, T.; Skoularigis, J.; Xanthopoulos, A. Hypertensive Heart Failure. Encyclopedia. Available online: https://encyclopedia.pub/entry/51311 (accessed on 23 July 2026).
Triposkiadis F, Sarafidis P, Briasoulis A, Magouliotis DE, Athanasiou T, Skoularigis J, et al. Hypertensive Heart Failure. Encyclopedia. Available at: https://encyclopedia.pub/entry/51311. Accessed July 23, 2026.
Triposkiadis, Filippos, Pantelis Sarafidis, Alexandros Briasoulis, Dimitrios E. Magouliotis, Thanos Athanasiou, John Skoularigis, Andrew Xanthopoulos. "Hypertensive Heart Failure" Encyclopedia, https://encyclopedia.pub/entry/51311 (accessed July 23, 2026).
Triposkiadis, F., Sarafidis, P., Briasoulis, A., Magouliotis, D.E., Athanasiou, T., Skoularigis, J., & Xanthopoulos, A. (2023, November 08). Hypertensive Heart Failure. In Encyclopedia. https://encyclopedia.pub/entry/51311
Triposkiadis, Filippos, et al. "Hypertensive Heart Failure." Encyclopedia. Web. 08 November, 2023.
Copy Citation
Hypertension (HTN) is the leading cause of cardiovascular disease and premature death worldwide, which largely surpasses other important factors of mortality such as smoking and metabolic diseases. HTN is the most important risk factor for heart failure (HF) development, with recent evidence indicating that HTN is present in 76% of incident HF cases, and the lifetime risk of HF is almost twice as high in people with HTN as in those with normal blood pressure (BP).
heart failure
hypertension
ejection fraction
autonomic imbalance
1. Introduction
Hypertension (HTN) is the leading cause of cardiovascular disease and premature death worldwide, which largely surpasses other important factors of mortality such as smoking and metabolic diseases [1][2][3]. HTN is the most important risk factor for heart failure (HF) development, with recent evidence indicating that HTN is present in 76% of incident HF cases [4], and the lifetime risk of HF is almost twice as high in people with HTN as in those with normal blood pressure (BP) [5][6]. From a pathophysiological standpoint, HTN causes left ventricular (LV) hypertrophy (LVH), fibrosis, and structural alterations of large and small arteries (microvascular disease) [7]. Further, several epidemiological studies have revealed the association between HTN and coronary artery disease (CAD), a major HF risk factor [7]. In this regard, in the INTERHEART study, 25% of the population-attributable risk of a myocardial infarction could be accounted for by HTN [8].
Despite overwhelming epidemiological evidence, the contribution of HTN to HF development has been undermined in current clinical practice. This is due to the fact that approximately half of HF patients have been labeled as suffering from HF with preserved left ventricular (LV) ejection fraction (EF) (HFpEF), with HTN, obesity, and diabetes mellitus (DM) being considered virtually equally responsible for its development [9]. However, this is inaccurate, since HTN is by far the most frequent and devastating morbidity present in HFpEF, with its prevalence reaching 80% in the Get With the Guidelines (GWTG) initiative [10], and 90% or more in large randomized clinical trials testing the effectiveness of medical treatment in HFpEF [11][12][13][14]. Further, HF development in obesity or DM is rare in the absence of HTN or CAD [15][16][17][18], whereas HTN often causes HF per se [19]. Finally, unlike HTN, for most major comorbidities present in HFpEF including anemia, chronic kidney disease, pulmonary disease, DM, atrial fibrillation (AF), sleep apnea, and depression, it is unknown whether they precede HF or result from it [20][21], and combinations of morbidities (multimorbidity) might occur randomly because the individual component conditions are common. Due to these drawbacks, comorbidity was recently redefined as the accumulation of additional morbidities to an index morbidity (a specific morbidity under consideration) over an individual’s lifetime [22]. This approach is of practical importance because it is crucial to discerning the settled mechanisms leading to multimorbidity, as chains of disease causation might be separated in time (e.g., antecedent HTN may give rise to CAD and myocardial infarction, which in turn can later lead to HF complicated by AF) [21].
2. Pathophysiology of Hypertension
The pathophysiological mechanisms responsible for HTN are complex, and act on a genetic background. Further, the probability of developing HTN increases with aging, due to the progressive stiffening of the arterial wall caused by, among other factors, slowly developing changes in vascular collagen and increases in atherosclerosis [23].
The mosaic theory of hypertension, which has prevailed to the present day, proposes that HTN pathophysiology is multifaceted. Accordingly, HTN is caused by multiple factors, including genetics, environment, adaptive, neural, mechanical, and hormonal perturbations, which intertwine to elevate BP [24][25]. Over the years, the Mosaic paradigm has been modified, and new concepts such as oxidative stress, inflammation, sodium homeostasis, and the microbiota have arisen, giving rise to further refinements of the ,osaic theory [26].
One of the recently proposed pathways for BP elevation involves the gut microbiota. The microbiota and the brain (i.e., the microbiota–gut–brain axis) communicate via various routes including the immune system, the vagus, and the enteric nervous system. Many factors can influence microbiota composition in early life, including infection, type of birth delivery, antibiotics, nutritional provision, environmental stressors, and host genetic factors. On the other hand, microbial diversity decreases with aging [27].
There is compelling evidence that shifts in gut microbiota play a key role in BP regulation. In this regard, intestinal bacteria synthesize metabolites, the most important being short-chain fatty acids (SCFAs), vasoactive hormones, trimethylamine (TMA) and trimethylamine N-oxide (TMAO) and uremic toxins, such as indoxyl sulfate (IS) and p-cresyl sulfate (PCS) [28]. Microbiota derangements are causally associated with HTN [29]. Hypertensive stimuli (stress, diet, salt, maternal factors, environmental toxins, etc.) significantly impact the microbiota-gut-brain axis and influence ANS brain regions to affect sympathetic, endocrine, and immune pathways [30]. Chronic overactivation of immune cells either directly or through the gut microbiota ultimately produces chronic inflammation and HTN. T cells are central to the immune responses underlying HTN, as activated T cells infiltrate tissues and produce cytokines including interleukin 17A, which promote renal and vascular dysfunction as well as end-organ damage, thereby leading to HTN [31].
In the gut itself, hypertensive stimuli cause gut microbial dysbiosis, gut barrier weakening, and inflammation [30]. Weakened barrier function allows previously excluded gut contents (e.g., bacteria and their metabolites) to come in contact with the immune system and generate inflammation, which in turn exacerbates leakiness and inflammation and recruits pro-inflammatory bone marrow progenitor cells to the gut generating a vicious cycle. The factors crossing the weakened gut barrier and inflammatory mediators generated in the gut reach the brain via the circulation and contribute to the development of local (neuroinflammation) and systemic inflammation [32].
Finally, there is bidirectional interaction between the microbiota and the renin angiotensin system (RAS), a major determinant of BP levels. Gut bacteria and their metabolites modulate gastrointestinal and systemic RAS, and at the same time, changes in the intestinal habitat caused by alterations in RAS may shape microbiota metabolic activity and composition [33]. Thus, an inflammatory milieu developed by hypertensive stimuli precedes, and is causally related to HTN development regardless of the presence or absence of other morbidities, rendering the theory of multimorbidity-induced inflammation leading to HFpEF obsolete [34]. It comes, therefore, as no surprise that HFpEF in the absence of HTN is rare [35].
The effect of the aforementioned perturbations on BP levels is also affected by genetic factors. Several genes have been identified to play a role in the pathophysiology of HTN, including those involved in the renin–angiotensin–aldosterone system (RAAS), catecholamine/adrenergic system, renal kallikrein–kinin system, epithelial sodium channel, adducin, and those involving lipoprotein metabolism, hormone receptors, and growth factors [36]. Genome-wide association studies (GWAS) have exposed more than 100 variants associated with blood pressure in the general population [37], and some studies have reported genes associated with hypertensive heart disease’s complications such as cardiomyopathy and HF [38]. Lastly, post-genomic biomarkers, from the emerging fields of transcriptomics, proteomics, glycomics, and lipidomics, have provided new insights into the molecular underpinnings of hypertension [36].
3. From Hypertension to Hypertensive Heart Failure
HTN is characterized by chronic LV pressure overload and increased intravascular volume, both affecting the LV structure and function [39]. Nevertheless, the conventional concept, described more than 120 years ago by William Osler, has been that HTN leads to concentric LV hypertrophy (LVH), which in turn is followed by eccentric LVH and HF [40]. In this regard, Messerli et al. proposed that HHF develops in four stages: [41] (a) stage I: isolated LV diastolic dysfunction with no LV hypertrophy; (b) stage II: LV diastolic dysfunction with concentric LVH; (c) stage III: clinical HF (dyspnea and pulmonary edema) with concentric LVH; and (d) stage IV: eccentric LVH with HF and reduced ejection fraction.
However, doubt has been raised that the previously mentioned sequence of events is typical in hypertensives, based on the following arguments: (a) concentric LVH may not be the most frequent geometric pattern, and is less commonly seen than eccentric LVH in studies enrolling hypertensive subjects [42]; (b) the transition from concentric LVH to eccentric LVH is uncommon in the absence of CAD [43]; (c) the risk varies by LV geometric pattern, with eccentric and concentric LVH predisposing individuals to HFrEF and HFpEF, respectively [44].
However, recent studies have provided compelling evidence that cardiac remodeling is a dynamic process, with phenotypic transitions occurring frequently regardless of the presence or absence of CAD. For example, in the Swedish Heart Failure Registry (SwedeHF), the proportion of patients with HFpEF (approximately 70% were hypertensive) that worsened during follow-up (from HFpEF to HF with mid-range LVEF [HFmrEF] or HF with reduced LVEF [HFrEF]) was 31.2% in the absence of baseline CAD, 33.1% in the presence of baseline CAD, and 57.2% in the case of interim CAD [45]. Similar were the findings of another study including 1082 patients (approximately 70% were hypertensive) admitted to hospital due to decompensated HFpEF (LVEF > 50% at the first LVEF assessment at discharge). At LVEF reassessment within 6 months in the outpatient setting, 758 patients (70%) had an LVEF > 50%, 138 patients (13%) had an LVEF of 40–49% (HFmrEF), and 186 patients (17%) had an LVEF of <40% (HFrEF) [46]. In addition, antihypertensive treatment attenuates or even reverses cardiac remodeling [47][48].
The port of entry in the HF spectrum (HHF entry phenotype) depends on (a) HTN severity, duration and antihypertensive treatment effectiveness; (b) the balance between LV pressure and LV volume overload; (c) the coexistence of morbidities such as obesity, DM, and CAD that preexist and/or modify LVH development; and (d) disease modifiers (age, sex, genes, other). The eventual HHF phenotype results from transitions across the HF spectrum, whose direction depends on disease severity and antihypertensive treatment, which shift towards the lower end of upper end of the HF spectrum.
4. Cardiac Autonomic Imbalance
HTN evolves over the lifespan, from predominant sympathetic nervous system (SNS)-driven HTN with elevated mean BP in early and mid-life to a late-life phenotype of increasing systolic and falling diastolic BP, associated with increased arterial stiffness and aortic pulsatility [49]. However, growing evidence indicates that the SNS is also capable of modulating arterial stiffness independently of prevailing hemodynamics and vasomotor tone [50][51]. The contribution of arterial stiffness to HTN and HF development is high in the elderly and patients with CKD [49][52].
The autonomic dysregulation observed in HTN escalates in HHF. SNS overactivity has long been appreciated as a compensatory mechanism initially supporting the failing heart, which, however, in the long term, triggers a sequence of unfavorable remodeling processes, causing HF progression and the occurrence of major cardiovascular events [53][54].
The adverse cardiac effects of SNS overactivity in HF have been predominantly studied in dilated LV with eccentric LVH, in which it manifests as an increase in norepinephrine spillover from the cardiac sympathetic endings, leading to chronic β-adrenergic receptor (AR) hyperstimulation and maladaptive GRK2 upregulation (GPCR kinase 2) [55], thereby promoting β-AR down-regulation, cardiac hypertrophy, and myocyte apoptosis. Further, GRK2 recruits β-arrestin, which then competes with G-proteins for interaction with the β-AR and limits their activation [56]. The chronic SNS overactivity In HF with eccentric LVH has been attributed to several neurogenic disturbances acting in concert [57].
SNS overactivity is also present in HF with nondilated hearts and lack of eccentric LVH. In this regard, earlier studies suggested that the SNS overdrive which is present in essential HTN is augmented in HF without eccentric LVH [58][59]. These findings have also been confirmed in recent HFpEF studies, in which most patients were hypertensive. Kaye et al. evaluated 14 healthy volunteers and 20 HFpEF patients (65% hypertensive), and found systemic sympathoexcitation in HFpEF patients, as indicated by the increased plasma arterial norepinephrine concentration and plasma levels of dihydroxyphenylglycol (a major intraneuronal metabolite of recaptured norepinephrine) compared with normal controls [60]. Seo et al., using iodine-123-labeled metaiodobenzylguanidine (123I-MIBG) single-photon emission computed tomography (SPECT) imaging in 148 patients admitted for acute decompensated nonischemic HFpEF (91% hypertensive) [61], observed that during a mean follow-up period of 2.4 ± 1.6 years, those with a high total defect score (TDS) levels had a significantly greater risk of cardiac events than those with middle or low TDS levels.
In contrast to SNS overactivity, there is attenuation of the parasympathetic nervous system (PNS) activity and its physiological effects in HF [62], including the PNS-mediated anti-inflammatory reflex [63]. The vagus nerve also plays a crucial role in this reflex, providing the afferent and efferent pathways which underly the communication between the brain and peripheral organs, including the heart [63][64]. Vagal sensory afferents are activated by proinflammatory cytokines in peripheral tissues and convey the signal to the brain. Subsequently this signal causes the release of acetylcholine from vagal efferents into the reticuloendothelial system, which inhibits proinflammatory cytokine synthesis and release. Thus, vagal withdrawal contributes to the creation of the inflammatory milieu in HF [65].
5. Treatment of Hypertensive Heart Failure
In patients with HF, treatment of HTN has been unanimously recommended, but BP targets have been inappropriately defined [66]. However, as RAAS and SNS overactivity play key roles in HTN and HF as well in the development of major complications such as kidney dysfunction, their inhibition is of outmost importance for the management of HHF [67]. Further, another class of agents, the sodium glucose cotransporter 2 inhibitors (SGLT-2i), which have proved tremendously effective in HF management, have a complex pleiotropic mechanism of action, including a reduction of neurohormonal overactivity [68].
5.1. Blood Pressure Targets
In the major outcome trials of hypertension, comparison of BP reduction with antihypertensive medications against placebo or no treatment, incident HF was the outcome that showed the largest intergroup risk reductions [69][70][71]. Effective HTN treatment in HF should target not only average BP, but the time in therapeutic range pressure range (TTR) as well.
5.1.1. Target Average Blood Pressure
It has been suggested that there is a J-shaped curve describing an inverse relationship between BP and cardiovascular complications, and that this association is more pronounced in patients with preexisting CAD, HTN, or LVH [72]. However, several lines of evidence dispute the presence of such a curve.
The target BP which should be pursued with medical treatment was evaluated in the pivotal SPRINT (Systolic Blood Pressure Intervention Trial), which demonstrated that among patients at high risk for cardiovascular events in the absence of DM, targeting a systolic BP < 120 mmHg (intensive treatment group; mean systolic BP at one year 121.4 mmHg), as compared with < 140 mmHg (standard treatment group; mean systolic BP at one year 136.2 mmHg), decreases the incidence of the primary outcome (myocardial infarction, other acute coronary syndromes, stroke, HF, or death from cardiovascular causes) [73]. Nevertheless, analyses comparing the effects of intensive and standard BP treatment in the ACCORD (Action to Control Cardiovascular Risk in Type 2 Diabetes) trial showed that the diabetic patients who received standard glycemic therapy and intensive BP control had benefits similar to those seen in SPRINT [74][75].
The results of SPRINT have been confirmed in several subsequent investigations. which demonstrated a significant and direct dose–response between systolic BP levels and ischemic heart disease risk across all systolic BP exposure values (100–200 mmHg), without evidence for a J-shaped curve [76][77].
According to the 2018 ESC/ESH guidelines, in hypertensive patients with HF, BP-lowering treatment should be considered if an individual’s BP is ≥140/90 mmHg, and systolic BP should be lowered to a range of 120–130 mmHg [7].
5.1.2. Time in Therapeutic Range
Although BP is a continuous and dynamic variable, a single or average BP value has been used for BP monitoring in clinical practice and HTN studies. To overcome this limitation, it has been recommended that doctors should pursue every point of BP monitoring instead of measuring a single value of BP [78]. In this regard, the term TTR was introduced, which expresses the percentage of BP measurements recorded within a certain window (e.g., TTR for BP window 120–140 mmHg) and reflects, therefore, the prevailing BP during the follow-up period and the magnitude of BP variability [79] (Table 1). The importance of TTR in HTN management was documented in a study including 371,996 hypertensive patients, in which the mortality rate increased from the most consistently controlled quartile (>75% in TTR) towards the less consistently controlled quartiles [79]. Likewise, a secondary analysis of the TOPCAT (Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist) trial, in which the TTR was calculated with the target range of systolic BP defined as 110 to 130 mmHg [80], a greater time in the systolic BP target range was associated with a decreased risk of cardiovascular outcomes and mortality events beyond BP level, especially among younger patients. Finally, in a recent post hoc analysis of HHF patients both from TOPCAT and BEST (Beta-Blocker Evaluation of Survival Trial) showed a linear relationship between TTR and the primary outcome (cardiovascular death or HF hospitalization), and similar patterns were observed in the individual trials [81]. Moreover, sensitivity analyses redefined target range as 110 to 130 mmHg for systolic BP or 70 to 80 mmHg for diastolic BP.
Table 1. Example illustrating the importance of time in therapeutic range (TTR) in hypertension management. Although the average systolic blood pressure (BP) achieved with treatment is similar among the three patients, the TTR (target range for systolic BP: 110–130 mmHg) significantly differs.
| Systolic BP | Patient A | Patient B | Patient C |
|---|---|---|---|
| Measurement 1 | 135 | 125 | 115 |
| Measurement 2 | 125 | 125 | 120 |
| Measurement 3 | 135 | 120 | 125 |
| Measurement 4 | 140 | 120 | 120 |
| Measurement 5 | 100 | 140 | 125 |
| Measurement 6 | 100 | 100 | 135 |
| Measurement 7 | 125 | 125 | 115 |
| Measurement 8 | 135 | 115 | 125 |
| Measurement 9 | 135 | 140 | 135 |
| Measurement 10 | 105 | 100 | 120 |
| Average BP (mmHg) | 123.5 | 121 | 123. 5 |
| Time to target (%) | 30% (3/10) | 60% (6/10) | 80% (8/10) |
These BP targets may also be applied in elderly HHF patients, as well as in those with coexistent DM [82] or kidney disease [83].
5.2. Medications
The drugs used in the management of HF, except for diuretics, have both cardioprotective and blood pressure-lowering properties (Table 2).
Table 2. Therapeutic targets and medications in the management of hypertensive heart failure.
| Congestion | Blood Pressure Lowering and Cardioprotection |
|
|---|---|---|
bumetanide) |
|
|
| Blood Pressure Targets Systolic BP 110–130 mmHg Time in therapeutic range > 75 % |
Comments: 1. Additional use of thiazide diuretics or carbonic anhydrase inhibitors in cases with diuretic resistance 2. Thiazide diuretics may be considered in decongested patients instead of loop diuretics for blood pressure control. |
Comments: 1. Besides effectively lowering BP, all the aforementioned classes of antihypertensives are cardioprotective. 2. Vasodilating beta-blockers with a favorable metabolic profile (e.g, carvedilol, nebivolol) may be preferable. 3. Beta-blockers are first-line agents in eccentric hypertrophy. 4. B-blockers should be used in selected patients with concentric hypertrophy (e.g., atrial fibrillation, angina, resistant hypertension). 5. The additional use of dihydropyridine calcium channel blockers should be considered when BP cannot be otherwise controlled. |
BP, blood pressure; ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; MRA, mineralocorticoid receptor antagonist.
5.2.1. RAAS Inhibitors (RAASi)
RAASi, that is, ACEi, ARBs, sacubtril/valsartan (ARNI), and MRAs, effectively reduce BP in hypertensive patients [84]. The aforementioned agents have also proved effective in HF regardless of the LVEF [85][86][87]. Interestingly, in a post hoc analysis of PARAGON-HF (Prospective Comparison of ARNI With ARB Global Outcomes in HF With Preserved Ejection Fraction), ARNI was found to be useful in treating apparent resistant hypertension in patients with HFpEF, even in those who continued to have an elevated BP, despite treatment with at least four antihypertensive drug classes, including an MRA [88].
Steroidal MRAs, such as spironolactone and eplerenone, have proved effective in patients with HF, resistant HTN, or CKD [89]. However, the associated risk of hyperkalemia and hormonal side effects limit their broad implementation, especially in patients with type 2 DM and moderate-to-advanced chronic CKD [90].
In addition to the beneficial effects on the heart, ACEi/ARB, ARNI may slow progression to kidney failure, and thus they are appropriate for initial therapy for managing HHF, especially, and coexistent DM [91]. Finerenone is a novel, nonsteroidal MRA which significantly reduces the risk of hard cardiovascular and kidney failure outcomes with a minimal risk of hyperkalemia in hypertensive patients with type two DM and a broad range of CKD, as demonstrated in the FIDELIO-DKD (Finerenone in Reducing Kidney Failure and Disease Progression in Diabetic Kidney Disease; 97% hypertensive patients) trial [92] and the FIGARO-DKD (Finerenone in Reducing Cardiovascular Mortality and Morbidity in Diabetic Kidney Disease; 96% hypertensive patients) trial [93][94].
Finally, as many patients with HHF and DM may manifest a resistant form of HTN, the use of ARNI instead of an ACEi/ARB together with an MRA may be considered. If BP remains elevated, antihypertensive drugs with complementary mechanisms of action and lack or minimal negative inotropy (e.g., diuretics or dihydropyridine calcium channel blockers) should be progressively added to lower BP [95]. Subgroup analyses of these trials also provided preliminary evidence that the efficacy and safety profile of finerenone was similar, irrespective of background therapy with other medications such as SGLT-2i (see below). In the ARTS-DN (Mineralocorticoid Receptor Antagonist Tolerability, Study-Diabetic Nephropathy) trial (823 patients [approximately 95% hypertensive] with type 2 DM and CKD, with an urine albumin-to-creatinine ratio 30 mg/g and an estimated glomerular filtration rate of, 30–90 mL/min per 1.73 m2 had a reduced daytime and night-time SBP according to 24 h ambulatory blood pressure monitoring [96].
5.2.2. Sodium Glucose Cotransporter 2 Inhibitors (SGLT-2i)
The SGLT-2i block the sodium–glucose cotransporter 2, which is located on the apical membrane of the proximal convoluted tubules, and therefore target glucose reabsorption [97][98]. The SGLT-2i also reduce blood pressure, but their underlying mechanism(s) is(are) incompletely understood. Potential explanations include (a) an osmotic effect of glucose, allowing more sodium and water to remain within the tubules, causing natriuresis [99]; (b) excess glucose and sodium excretion, leading to RAAS Modification [100]; and (c) attenuation of SNS overactivity [101][102].
Recent clinical trials have reported that SGLT-2i have a beneficial effect on BP, which is not just an acute effect of treatment initiation, but has a long-term impact on both systolic and diastolic BP [103]. Further, SGLT-2i significantly reduces the rate of cardiovascular events (including HF) and prevents the progression of renal dysfunction and CKD development in patients with or without DM already receiving optimal medical therapy [104]. However, the reduction in BP in these trials was “modest”, and not of a magnitude to fully account for the approximately 30–40% reduction in HF, end-stage kidney disease, or renal or cardiovascular mortality [105], providing further evidence in support of the pleiotropic effects of SGLT-2i [106].
In addition to HTN, SGLT-2i have proved tremendously effective in reducing morbidity and mortality in HF patients irrespective of LVEF and diabetic status [107][108]. Regarding the SGLT-2i effects in HF, a recent meta-analysis of 16 RCTs reported that SGLT-2i were associated with a statistically significant reduction in systolic BP of 1.68 mmHg, no significant change in diastolic BP, a 1.36 kg decrease in body weight, a 0.16% decrease in glycated hemoglobin level, a 1.36% increase in hematocrit, and no change in heart rate [109]. The mechanisms underlying the beneficial effects of SGLT-2i in HF have not been delineated. According to an interesting hypothesis, the SGLT-2i rebalance the reabsorption of Na+ coupled with glucose and restore renal O2 demand, diminishing neuroendocrine activation [98]. Alternatively, SGLT-2i, by alleviating inflammation and oxidative stress, down-regulate hepcidin, upregulate transferrin receptor protein 1, and reduce ferritin, the net result being an increase the levels of cytosolic Fe2+ available to mitochondria, thus enabling the synthesis of heme in erythroid precursors and ATP in cardiomyocytes [110].
5.2.3. Beta Adrenergic Receptor Blockers (BBs)
BBs lower BP as effectively as other major antihypertensive drugs and have solid documentation in preventing cardiovascular complications [111]. BBs can be broadly classified into (a) nonselective β-blockers (e.g., propranolol) with similar β1 and β2 activity (none of the β-blockers belonging to this class are indicated for HF); (b) β1-selective blockers with a higher affinity for β1-adrenoreceptors (metoprolol, bisoprolol, and nebivolol), preferred in patients with chronic obstructive pulmonary disease or mild asthma (nebivolol also facilitates nitric oxide release, causes vasodilation, and is preferred in patients with HTN); and (c) β-blockers with additional α-1-adrenoreceptor antagonism and consequent peripheral, vasodilation (carvedilol), preferred in patients with HTN or documented higher peripheral vascular resistance.
Although BBs have proved to be lifesaving in HHF with eccentric LVH [112], they have never been appropriately tested in HF with concentric LVH [113], presumably due to the misbelief that the latter is not associated with SNS overactivity, which dominated for decades [114]. Moreover, it has been argued that pharmacological heart rate lowering in HHF patients with concentric LVH may have adverse effects on hemodynamics, exercise capacity, and outcomes [115]. Nevertheless, these complications often occur in HHF patients [35][116] and it comes, therefore, as no surprise that in recent large HFpEF trials, approximately 80% of study participants were treated with BBs [12][13][14].
5.2.4. Diuretics
Loop diuretics, which inhibit the Na+/K+/2Cl− symporter at the ascending limp loop of Henle, have the most potent diuretic effect, promote excretion of sodium and chloride (and potassium, albeit to a lesser extent than thiazides), and form the backbone of diuretic therapy in congestive HF [117]. Adequate dosing with sufficient plasma levels is pivotal as renal perfusion is often reduced in HF, resulting in diminished secretion of loop diuretics.
Thiazide diuretics are most commonly used to treat HTN, although they can be adjuncts in HF management on top of loop diuretics in patients with diuretic resistance [118]. They inhibit the Na+/Cl− symporter in the distal convoluted tubule, leading to decreased sodium and water reabsorption. They may prescribed for patients with HHF following decongestion instead of loop diuretics due to (a) their efficacy and low side effect profile; (b) their synergistic effect when combined with other antihypertensive agents; and (c) their counteraction of salt and fluid retention caused by other antihypertensive agents [119].
5.2.5. Implementation of Medical Treatment
The optimal time frame from initiation of antihypertensive therapy to attaining the levels of BP control that influence cardiovascular outcomes is not well defined. Overall, a series of landmark trials in hypertensive patients and additional cardiovascular risk factors collectively support the prompt achievement of BP control, ideally within 1–3 months [120].
The main obstacles to up-titration of medications in HF patients include a low BP, impaired renal function, electrolyte imbalance, and drug intolerance, which are frequently encountered during optimization of HF medications. However, the presence of elevated BP in HHF as well as the renoprotective effects of finerenone, which rarely causes early hyperkalemia [121], allows ultra-fast up-titration of HF medications. Thus, treatment should start with the simultaneous use of sacubitril/valsartan, SGLT-2i and finerenone. In HFF patients with eccentric LVH, BBs should be started from the beginning, whereas in HHF patients with concentric LVH, BBs should be used in selected patients, as previously mentioned (Figure 1).
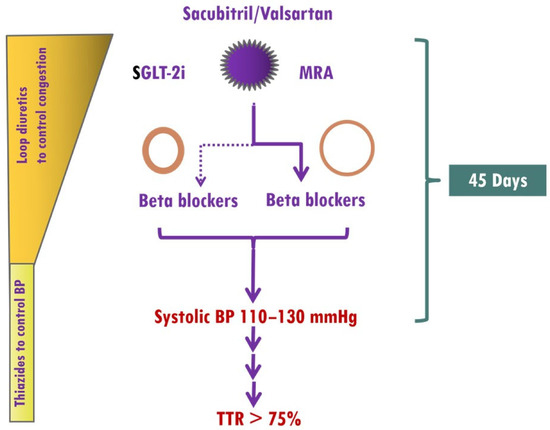
Figure 1. Implementation of medical treatment in hypertensive heart failure (HHF). The presence of elevated BP in HHF as well as the renoprotective effects of finerenone, which rarely causes early hyperkalemia, allows ultra-fast up-titration of HF medications. Treatment should start with the simultaneous use of sacubitril/valsartan, sodium glucose cotransporter 2 inhibitors (SGLT-2i), and mineralocorticoid receptor antagonists (MRAs, preferably finerenone). In HFF patients with eccentric left ventricular hypertrophy (LVH), BBs (preferably vasodilatory) should be started from the beginning, whereas in HHF patients with concentric LVH, BBs should be considered in those with atrial fibrillation, coronary artery disease, or resistant hypertension. A target systolic blood pressure of 110–130 mmHg should be achieved within 45 days, and thereafter, systolic blood pressure should be within the target range.
A majo© iss©e impeding ©he implementation of optimal medical treatment of HTN and HF is poor patient adherence to the prescribed drugs [122]. In this regard, treatment with fixed-dose combination pills may improve patient’s adherence and cardiovascular outcomes. An observational retrospective study of three Italian Local Health Units demonstrated that only half of patients prescribed atorvastatin–perindopril–amlodipine as a free combination were adherent to the medical therapy, whereas it was estimated that 69,542 hypertensive patients could be eligible for a fixed dose of atorvastatin–perindopril–amlodipine during 2014 [123]. An open-label multicenter study including 356 patients with HTN and/or CAD from 17 centers in Poland between January 2015 and August 2016, showed that a fixed-dose combination of bisoprolol and aspirin was associated with excellent or good (≥76%) adherence in 98.3% and 98.0% of patients based on pill counts and patients ‘diaries, respectively. A significant decrease was also observed in mean systolic BP, mean diastolic BP, and heart rate over the 3-month period (all p < 0.001) [124]. Many new drug combinations have been approved over the last few years, allowing the development of new single-pill formulations. The use of single-pill drugs seems to be optimal in the treatment of patients with HHF and concomitant diseases such as hyperlipidemia, AF, and DM.
5.3. Exercise
Exercise training is internationally recommended for patients with HF, regardless of LVEF [125]. Optimal exercise prescription enhances exercise capacity, improves quality of life, and reduces hospitalizations and mortality in HF patients. Specifically, physical activity promotes cardiovascular health by [126][127][128][129] (a) attenuating the negative effect of many established risk factors for cardiovascular disease (cholesterol, insulin sensitivity, body mass index, cholesterol, BP, sleep apnea); (b) directly influencing the cardiovascular system (increasing nitric oxide bioavailability, enhancing endothelial function, favoring arterial wall remodeling consisting of increased diameter and dilation capacity, decreasing coronary and wall thickness, developing coronary collateral vessels, and stabilizing coronary atheromatic plaque); (c) restoring the ANS balance and protecting against fetal arrhythmias; and (d) exhibiting antithrombotic and anti-inflammatory effects. Importantly, exercise interventions based on aerobic, combined, or isometric exercise are probably the most important currently available means to decrease arterial stiffness in adults with HTN [130].
5.4. Emerging Treatments
As the gut microbiota and their metabolites may contribute to HTN development, they represent a reasonable treatment target [131]. Since the gut microbiota is a complex and highly interactive system for the treatment of HTN, a “one-size fits all” approach using a metabolite or bacterial strain is unlikely to restore the perturbed metabolic activity associated with intestinal dysbiosis in HTN. Instead, probiotic supplementation using diverse bacterial strains producing or reducing the production of certain metabolites to elicit an antihypertensive effect is a potential treatment strategy for HTN management. There is evidence to suggest that intervention with probiotics could lower BP, modify the intestinal flora to increase the abundance of beneficial bacteria, and regulate intestinal microbial metabolites such as trimethylamine oxide, short-chain fatty acids, and polyphenols [132]. A recent umbrella meta-analysis which assessed the pooled effect size of 14 meta-analyses with 15,494 participants indicated significant decreases in both systolic and diastolic BP following probiotics supplementation. Greater effects on SBP were revealed in trials with a mean age of >50 years and the duration of intervention ≤10 Weeks [133]. DBP was also more reduced in studies with a dosage of ≥1010 colony-forming units (CFUs), and SBP was decreased in patients with HTN or DM, when analyzing mean difference. Gut microbiota science has far-reaching clinical implications for the diagnosis, prognosis, and monitoring of cardiovascular diseases in general, and HHF in particular. Dietary supplementation remains the safest probable method to improve the gut microbiota and its associated detrimental cardiovascular effects.
As increased SNS and decreased PNS activities are associated with HTN and HF, the neuromodulatory therapies of renal denervation (RDN) and vagus nerve stimulation (VNS) have received recent attention [134]. Catheter-based RDN can be used as an adjunct treatment option in uncontrolled resistant HTN despite best efforts toward lifestyle and pharmacological interventions, as well as in patients intolerant to antihypertensive medications in the long term [135]. A shared decision making process is a key feature, and preferably includes a patient who is well informed on the benefits and limitations of the procedure. Further studies are necessary to answer questions related to the impact of BP lowering with RDN on clinical outcomes and potential clinical indications beyond RDN. As the vagus nerve plays a critical role in the inflammatory reflex [136], VNS could attenuate the proinflammatory state known to be a key pathological mechanism in HTN and HHF, and attenuate cardiac remodeling. It is of interest that VNS can be performed in a less invasive manner via transcutaneous stimulation of the auricular branch of the vagus nerve, using the tragus of the ear as an anatomical landmark. In a recent study, HFpEF patients (96% hypertensive) were randomized to either active (tragus) or sham (earlobe) low-level transcutaneous vagus nerve stimulation (20 Hz, 1 mA below discomfort threshold), for 1 h daily for 3 months [137]. After three months, diastolic function improved and inflammatory markers decreased.
References
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Muntner, P.; Whelton, P.K.; He, J. Global burden of hypertension: Analysis of worldwide data. Lancet 2005, 365, 217–223.
- Murray, C.J.; Lopez, A.D. Measuring the global burden of disease. N. Engl. J. Med. 2013, 369, 448–457.
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237.
- Conrad, N.; Judge, A.; Tran, J.; Mohseni, H.; Hedgecott, D.; Crespillo, A.P.; Allison, M.; Hemingway, H.; Cleland, J.G.; McMurray, J.J.V.; et al. Temporal trends and patterns in heart failure incidence: A population-based study of 4 million individuals. Lancet 2018, 391, 572–580.
- Rapsomaniki, E.; Timmis, A.; George, J.; Pujades-Rodriguez, M.; Shah, A.D.; Denaxas, S.; White, I.R.; Caulfield, M.J.; Deanfield, J.E.; Smeeth, L.; et al. Blood pressure and incidence of twelve cardiovascular diseases: Lifetime risks, healthy life-years lost, and age-specific associations in 1.25 million people. Lancet 2014, 383, 1899–1911.
- Tromp, J.; Paniagua, S.M.A.; Lau, E.S.; Allen, N.B.; Blaha, M.J.; Gansevoort, R.T.; Hillege, H.L.; Lee, D.E.; Levy, D.; Vasan, R.S.; et al. Age dependent associations of risk factors with heart failure: Pooled population based cohort study. BMJ 2021, 372, n461.
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104.
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952.
- Nagueh, S.F. Heart failure with preserved ejection fraction: Insights into diagnosis and pathophysiology. Cardiovasc. Res. 2021, 117, 999–1014.
- Steinberg, B.A.; Zhao, X.; Heidenreich, P.A.; Peterson, E.D.; Bhatt, D.L.; Cannon, C.P.; Hernandez, A.F.; Fonarow, G.C.; Get with the Guidelines Scientific Advisory Committee and Investigators. Trends in patients hospitalized with heart failure and preserved left ventricular ejection fraction: Prevalence, therapies, and outcomes. Circulation 2012, 126, 65–75.
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 2014, 370, 1383–1392.
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620.
- Anker, S.D.; Butler, J.; Packer, M. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. Reply. N. Engl. J. Med. 2022, 386, e57.
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098.
- Dandamudi, S.; Slusser, J.; Mahoney, D.W.; Redfield, M.M.; Rodeheffer, R.J.; Chen, H.H. The prevalence of diabetic cardiomyopathy: A population-based study in Olmsted County, Minnesota. J. Card. Fail. 2014, 20, 304–309.
- Ren, J.; Wu, N.N.; Wang, S.; Sowers, J.R.; Zhang, Y. Obesity cardiomyopathy: Evidence, mechanisms, and therapeutic implications. Physiol. Rev. 2021, 101, 1745–1807.
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Albanese, G.; Di Martino, A.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Sasso, F.C. The Diabetic Cardiomyopathy: The Contributing Pathophysiological Mechanisms. Front. Med. 2021, 8, 695792.
- Triposkiadis, F.; Xanthopoulos, A.; Bargiota, A.; Kitai, T.; Katsiki, N.; Farmakis, D.; Skoularigis, J.; Starling, R.C.; Iliodromitis, E. Diabetes Mellitus and Heart Failure. J. Clin. Med. 2021, 10, 3682.
- Giamouzis, G.; Xanthopoulos, A.; Papamichalis, M.; Chroub-Papavaiou, A.N.; Pantziou, A.; Simou, A.; Dimos, A.; Bourazana, A.; Skoularigis, J.; Triposkiadis, F. Relative contribution of risk factors/co-morbidities to heart failure pathogenesis: Interaction with ejection fraction. ESC Heart Fail. 2020, 7, 4399–4403.
- Triposkiadis, F.; Starling, R.C.; Boudoulas, H.; Giamouzis, G.; Butler, J. The cardiorenal syndrome in heart failure: Cardiac? renal? syndrome? Heart Fail. Rev. 2012, 17, 355–366.
- Triposkiadis, F.; Giamouzis, G.; Parissis, J.; Starling, R.C.; Boudoulas, H.; Skoularigis, J.; Butler, J.; Filippatos, G. Reframing the association and significance of co-morbidities in heart failure. Eur. J. Heart Fail. 2016, 18, 744–758.
- Kuan, V.; Denaxas, S.; Patalay, P.; Nitsch, D.; Mathur, R.; Gonzalez-Izquierdo, A.; Sofat, R.; Partridge, L.; Roberts, A.; Wong, I.C.K.; et al. Identifying and visualising multimorbidity and comorbidity patterns in patients in the English National Health Service: A population-based study. Lancet Digit. Health 2023, 5, e16–e27.
- Triposkiadis, F.; Xanthopoulos, A.; Lampropoulos, K.; Briasoulis, A.; Sarafidis, P.; Skoularigis, J.; Boudoulas, H. Aortic Stiffness: A Major Risk Factor for Multimorbidity in the Elderly. J. Clin. Med. 2023, 12, 2321.
- Frohlich, E.D.; Dustan, H.P.; Bumpus, F.M. Irvine H. Page: 1901–1991. The celebration of a leader. Hypertension 1991, 18, 443–445.
- Harrison, D.G. The mosaic theory revisited: Common molecular mechanisms coordinating diverse organ and cellular events in hypertension. J. Am. Soc. Hypertens. 2013, 7, 68–74.
- Harrison, D.G.; Coffman, T.M.; Wilcox, C.S. Pathophysiology of Hypertension: The Mosaic Theory and Beyond. Circ. Res. 2021, 128, 847–863.
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013.
- Tokarek, J.; Budny, E.; Saar, M.; Kucmierz, J.; Mlynarska, E.; Rysz, J.; Franczyk, B. Does the Composition of Gut Microbiota Affect Hypertension? Molecular Mechanisms Involved in Increasing Blood Pressure. Int. J. Mol. Sci. 2023, 24, 1377.
- O’Donnell, J.A.; Zheng, T.; Meric, G.; Marques, F.Z. The gut microbiome and hypertension. Nat. Rev. Nephrol. 2023, 19, 153–167.
- Richards, E.M.; Li, J.; Stevens, B.R.; Pepine, C.J.; Raizada, M.K. Gut Microbiome and Neuroinflammation in Hypertension. Circ. Res. 2022, 130, 401–417.
- Madhur, M.S.; Elijovich, F.; Alexander, M.R.; Pitzer, A.; Ishimwe, J.; Van Beusecum, J.P.; Patrick, D.M.; Smart, C.D.; Kleyman, T.R.; Kingery, J.; et al. Hypertension: Do Inflammation and Immunity Hold the Key to Solving this Epidemic? Circ. Res. 2021, 128, 908–933.
- Bostick, J.W.; Schonhoff, A.M.; Mazmanian, S.K. Gut microbiome-mediated regulation of neuroinflammation. Curr. Opin. Immunol. 2022, 76, 102177.
- Jaworska, K.; Koper, M.; Ufnal, M. Gut microbiota and renin-angiotensin system: A complex interplay at local and systemic levels. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G355–G366.
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271.
- Xanthopoulos, A.; Triposkiadis, F.; Starling, R.C. Heart failure with preserved ejection fraction: Classification based upon phenotype is essential for diagnosis and treatment. Trends Cardiovasc. Med. 2018, 28, 392–400.
- Adua, E. Decoding the mechanism of hypertension through multiomics profiling. J. Hum. Hypertens. 2023, 37, 253–264.
- Seidel, E.; Scholl, U.I. Genetic mechanisms of human hypertension and their implications for blood pressure physiology. Physiol. Genom. 2017, 49, 630–652.
- Masenga, S.K.; Kirabo, A. Hypertensive heart disease: Risk factors, complications and mechanisms. Front. Cardiovasc. Med. 2023, 10, 1205475.
- Nadruz, W. Myocardial remodeling in hypertension. J. Hum. Hypertens. 2015, 29, 1–6.
- Osler, W. The Principles and Practice of Medicine; Appleton: New York, NY, USA, 1892.
- Messerli, F.H.; Rimoldi, S.F.; Bangalore, S. The Transition From Hypertension to Heart Failure: Contemporary Update. JACC Heart Fail. 2017, 5, 543–551.
- Cuspidi, C.; Sala, C.; Negri, F.; Mancia, G.; Morganti, A.; Italian Society of Hypertension. Prevalence of left-ventricular hypertrophy in hypertension: An updated review of echocardiographic studies. J. Hum. Hypertens. 2012, 26, 343–349.
- Drazner, M.H. The progression of hypertensive heart disease. Circulation 2011, 123, 327–334.
- Velagaleti, R.S.; Gona, P.; Pencina, M.J.; Aragam, J.; Wang, T.J.; Levy, D.; D’Agostino, R.B.; Lee, D.S.; Kannel, W.B.; Benjamin, E.J.; et al. Left ventricular hypertrophy patterns and incidence of heart failure with preserved versus reduced ejection fraction. Am. J. Cardiol. 2014, 113, 117–122.
- Vedin, O.; Lam, C.S.P.; Koh, A.S.; Benson, L.; Teng, T.H.K.; Tay, W.T.; Braun, O.O.; Savarese, G.; Dahlstrom, U.; Lund, L.H. Significance of Ischemic Heart Disease in Patients with Heart Failure and Preserved, Midrange, and Reduced Ejection Fraction: A Nationwide Cohort Study. Circ. Heart Fail. 2017, 10, e003875.
- Yamanaka, S.; Sakata, Y.; Nochioka, K.; Miura, M.; Kasahara, S.; Sato, M.; Aoyanagi, H.; Fujihashi, T.; Hayashi, H.; Shiroto, T.; et al. Prognostic impacts of dynamic cardiac structural changes in heart failure patients with preserved left ventricular ejection fraction. Eur. J. Heart Fail. 2020, 22, 2258–2268.
- Milani, R.V.; Lavie, C.J.; Mehra, M.R.; Ventura, H.O.; Kurtz, J.D.; Messerli, F.H. Left ventricular geometry and survival in patients with normal left ventricular ejection fraction. Am. J. Cardiol. 2006, 97, 959–963.
- Lamirault, G.; Artifoni, M.; Daniel, M.; Barber-Chamoux, N.; Nantes University Hospital Working Group On, H. Resistant Hypertension: Novel Insights. Curr. Hypertens. Rev. 2020, 16, 61–72.
- Triposkiadis, F.; Xanthopoulos, A.; Butler, J. Cardiovascular Aging and Heart Failure: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 804–813.
- Holwerda, S.W.; Luehrs, R.E.; DuBose, L.; Collins, M.T.; Wooldridge, N.A.; Stroud, A.K.; Fadel, P.J.; Abboud, F.M.; Pierce, G.L. Elevated Muscle Sympathetic Nerve Activity Contributes to Central Artery Stiffness in Young and Middle-Age/Older Adults. Hypertension 2019, 73, 1025–1035.
- Nardone, M.; Floras, J.S.; Millar, P.J. Sympathetic neural modulation of arterial stiffness in humans. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H1338–H1346.
- Zern, E.K.; Ho, J.E.; Panah, L.G.; Lau, E.S.; Liu, E.; Farrell, R.; Sbarbaro, J.A.; Schoenike, M.W.; Pappagianopoulos, P.P.; Namasivayam, M.; et al. Exercise Intolerance in Heart Failure with Preserved Ejection Fraction: Arterial Stiffness and Aabnormal Left Ventricular Hemodynamic Responses During Exercise. J. Card. Fail. 2021, 27, 625–634.
- Floras, J.S.; Ponikowski, P. The sympathetic/parasympathetic imbalance in heart failure with reduced ejection fraction. Eur. Heart J. 2015, 36, 1974–1982.
- Gronda, E.; Dusi, V.; D’Elia, E.; Iacoviello, M.; Benvenuto, E.; Vanoli, E. Sympathetic activation in heart failure. Eur. Heart J. Suppl. 2022, 24, E4–E11.
- Bencivenga, L.; Palaia, M.E.; Sepe, I.; Gambino, G.; Komici, K.; Cannavo, A.; Femminella, G.D.; Rengo, G. Why Do We Not Assess Sympathetic Nervous System Activity in Heart Failure Management: Might GRK2 Serve as a New Biomarker? Cells 2021, 10, 457.
- Boussi, L.; Frishman, W.H. beta-Arrestin as a Therapeutic Target in Heart Failure. Cardiol. Rev. 2021, 29, 223–229.
- Floras, J.S. Alterations in the sympathetic and parasympathetic nnervous systems in heart failure. In Heart Failure: A Companion to Braunwald’s Heart Disease; Felker, M.G., Mann, D.L., Eds.; Elsevier: Toronto, ON, Canada, 2019; pp. 181–200.
- Grassi, G.; Seravalle, G.; Quarti-Trevano, F.; Dell’Oro, R.; Arenare, F.; Spaziani, D.; Mancia, G. Sympathetic and baroreflex cardiovascular control in hypertension-related left ventricular dysfunction. Hypertension 2009, 53, 205–209.
- Verloop, W.L.; Beeftink, M.M.; Santema, B.T.; Bots, M.L.; Blankestijn, P.J.; Cramer, M.J.; Doevendans, P.A.; Voskuil, M. A systematic review concerning the relation between the sympathetic nervous system and heart failure with preserved left ventricular ejection fraction. PLoS ONE 2015, 10, e0117332.
- Kaye, D.M.; Nanayakkara, S.; Wang, B.; Shihata, W.; Marques, F.Z.; Esler, M.; Lambert, G.; Mariani, J. Characterization of Cardiac Sympathetic Nervous System and Inflammatory Activation in HFpEF Patients. JACC Basic. Transl. Sci. 2022, 7, 116–127.
- Seo, M.; Yamada, T.; Tamaki, S.; Watanabe, T.; Morita, T.; Furukawa, Y.; Kawasaki, M.; Kikuchi, A.; Kawai, T.; Nakamura, J.; et al. Prognostic Significance of Cardiac (123)I-MIBG SPECT Imaging in Heart Failure Patients with Preserved Ejection Fraction. JACC Cardiovasc. Imaging 2022, 15, 655–668.
- Olshansky, B.; Sabbah, H.N.; Hauptman, P.J.; Colucci, W.S. Parasympathetic nervous system and heart failure: Pathophysiology and potential implications for therapy. Circulation 2008, 118, 863–871.
- Ramos-Martinez, I.E.; Rodriguez, M.C.; Cerbon, M.; Ramos-Martinez, J.C.; Ramos-Martinez, E.G. Role of the Cholinergic Anti-Inflammatory Reflex in Central Nervous System Diseases. Int. J. Mol. Sci. 2021, 22, 13427.
- Kittipibul, V.; Fudim, M. Tackling Inflammation in Heart Failure with Preserved Ejection Fraction: Resurrection of Vagus Nerve Stimulation? J. Am. Heart Assoc. 2022, 11, e024481.
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285.
- Pinho-Gomes, A.C.; Rahimi, K. Management of blood pressure in heart failure. Heart 2019, 105, 589–595.
- Singhania, N.; Bansal, S.; Mohandas, S.; Nimmatoori, D.P.; Ejaz, A.A.; Singhania, G. Role of renin-angiotensin-aldosterone system inhibitors in heart failure and chronic kidney disease. Drugs Context 2020, 9, 2020-7-3.
- Sano, M. A new class of drugs for heart failure: SGLT2 inhibitors reduce sympathetic overactivity. J. Cardiol. 2018, 71, 471–476.
- Moser, M.; Hebert, P.R. Prevention of disease progression, left ventricular hypertrophy and congestive heart failure in hypertension treatment trials. J. Am. Coll. Cardiol. 1996, 27, 1214–1218.
- Mancia Chairperson, G.; Kreutz Co-Chair, R.; Brunstrom, M.; Burnier, M.; Grassi, G.; Januszewicz, A.; Muiesan, M.L.; Tsioufis, K.; Agabiti-Rosei, E.; Algharably, E.A.E.; et al. 2023 ESH Guidelines for the management of arterial hypertension The Task Force for the management of arterial hypertension of the European Society of Hypertension Endorsed by the European Renal Association (ERA) and the International Society of Hypertension (ISH). J. Hypertens. 2023.
- Moser, M.; Gifford, R.W. Hypertension: Steps forward and steps backward. Arch. Intern. Med. 1993, 153, 1843–1846.
- Maeda, D.; Dotare, T.; Matsue, Y.; Teramoto, K.; Sunayama, T.; Tromp, J.; Minamino, T. Blood pressure in heart failure management and prevention. Hypertens. Res. 2023, 46, 817–833.
- Group, S.R.; Wright, J.T., Jr.; Williamson, J.D.; Whelton, P.K.; Snyder, J.K.; Sink, K.M.; Rocco, M.V.; Reboussin, D.M.; Rahman, M.; Oparil, S.; et al. A Randomized Trial of Intensive versus Standard Blood-Pressure Control. N. Engl. J. Med. 2015, 373, 2103–2116.
- Buckley, L.F.; Dixon, D.L.; Wohlford, G.F.t.; Wijesinghe, D.S.; Baker, W.L.; Van Tassell, B.W. Intensive Versus Standard Blood Pressure Control in SPRINT-Eligible Participants of ACCORD-BP. Diabetes Care 2017, 40, 1733–1738.
- Wright, J.T., Jr.; Whelton, P.K.; Johnson, K.C.; Snyder, J.K.; Reboussin, D.M.; Cushman, W.C.; Williamson, J.D.; Pajewski, N.M.; Cheung, A.K.; Lewis, C.E.; et al. SPRINT Revisited: Updated Results and Implications. Hypertension 2021, 78, 1701–1710.
- Blood Pressure Lowering Treatment Trialists, C. Pharmacological blood pressure lowering for primary and secondary prevention of cardiovascular disease across different levels of blood pressure: An individual participant-level data meta-analysis. Lancet 2021, 397, 1625–1636.
- Razo, C.; Welgan, C.A.; Johnson, C.O.; McLaughlin, S.A.; Iannucci, V.; Rodgers, A.; Wang, N.; LeGrand, K.E.; Sorensen, R.J.D.; He, J.; et al. Effects of elevated systolic blood pressure on ischemic heart disease: A Burden of Proof study. Nat. Med. 2022, 28, 2056–2065.
- Mancia, G.; Messerli, F.; Bakris, G.; Zhou, Q.; Champion, A.; Pepine, C.J. Blood pressure control and improved cardiovascular outcomes in the International Verapamil SR-Trandolapril Study. Hypertension 2007, 50, 299–305.
- Doumas, M.; Tsioufis, C.; Fletcher, R.; Amdur, R.; Faselis, C.; Papademetriou, V. Time in Therapeutic Range, as a Determinant of All-Cause Mortality in Patients with Hypertension. J. Am. Heart Assoc. 2017, 6, e007131.
- Huang, R.; Lin, Y.; Liu, M.; Xiong, Z.; Zhang, S.; Zhong, X.; Ye, X.; Huang, Y.; Zhuang, X.; Liao, X. Time in Target Range for Systolic Blood Pressure and Cardiovascular Outcomes in Patients with Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2022, 11, e022765.
- Chen, K.; Li, C.; Cornelius, V.; Yu, D.; Wang, Q.; Shi, R.; Wu, Z.; Su, H.; Yan, J.; Chen, T.; et al. Prognostic Value of Time in Blood Pressure Target Range Among Patients with Heart Failure. JACC Heart Fail. 2022, 10, 369–379.
- Kim, H.J.; Kim, K.I. Blood Pressure Target in Type 2 Diabetes Mellitus. Diabetes Metab. J. 2022, 46, 667–674.
- Burnier, M.; Damianaki, A. Hypertension as Cardiovascular Risk Factor in Chronic Kidney Disease. Circ. Res. 2023, 132, 1050–1063.
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-angiotensin-aldosterone system alterations. Circ. Res. 2015, 116, 960–975.
- Lund, L.H.; Claggett, B.; Liu, J.; Lam, C.S.; Jhund, P.S.; Rosano, G.M.; Swedberg, K.; Yusuf, S.; Granger, C.B.; Pfeffer, M.A.; et al. Heart failure with mid-range ejection fraction in CHARM: Characteristics, outcomes and effect of candesartan across the entire ejection fraction spectrum. Eur. J. Heart Fail. 2018, 20, 1230–1239.
- Solomon, S.D.; Vaduganathan, M.; Claggett, B.L.; Packer, M.; Zile, M.; Swedberg, K.; Rouleau, J.; Pfeffer, M.A.; Desai, A.; Lund, L.H.; et al. Sacubitril/Valsartan Across the Spectrum of Ejection Fraction in Heart Failure. Circulation 2020, 141, 352–361.
- Pfeffer, M.A.; Claggett, B.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; Gordeev, I.; et al. Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist (TOPCAT) trial. Circulation 2015, 131, 34–42.
- Jackson, A.M.; Jhund, P.S.; Anand, I.S.; Dungen, H.D.; Lam, C.S.P.; Lefkowitz, M.P.; Linssen, G.; Lund, L.H.; Maggioni, A.P.; Pfeffer, M.A.; et al. Sacubitril-valsartan as a treatment for apparent resistant hypertension in patients with heart failure and preserved ejection fraction. Eur. Heart J. 2021, 42, 3741–3752.
- Carey, R.M.; Calhoun, D.A.; Bakris, G.L.; Brook, R.D.; Daugherty, S.L.; Dennison-Himmelfarb, C.R.; Egan, B.M.; Flack, J.M.; Gidding, S.S.; Judd, E.; et al. Resistant Hypertension: Detection, Evaluation, and Management: A Scientific Statement From the American Heart Association. Hypertension 2018, 72, e53–e90.
- Gregg, L.P.; Navaneethan, S.D. Steroidal or non-steroidal MRAs: Should we still enable RAASi use through K binders? Nephrol. Dial. Transplant. 2023, 38, 1355–1365.
- Banerjee, D.; Winocour, P.; Chowdhury, T.A.; De, P.; Wahba, M.; Montero, R.; Fogarty, D.; Frankel, A.H.; Karalliedde, J.; Mark, P.B.; et al. Management of hypertension and renin-angiotensin-aldosterone system blockade in adults with diabetic kidney disease: Association of British Clinical Diabetologists and the Renal Association UK guideline update 2021. BMC Nephrol. 2022, 23, 9.
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229.
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263.
- Ruolin, L.; Lili, X.; Lin, C.; Song, L.; Yangang, W.; Bingzi, D. Cardiovascular-renal protective effect and molecular mechanism of finerenone in type 2 diabetic mellitus. Front. Endocrinol. 2023, 14, 1125693.
- Jia, G.; Sowers, J.R. Hypertension in Diabetes: An Update of Basic Mechanisms and Clinical Disease. Hypertension 2021, 78, 1197–1205.
- Agarwal, R.; Ruilope, L.M.; Ruiz-Hurtado, G.; Haller, H.; Schmieder, R.E.; Anker, S.D.; Filippatos, G.; Pitt, B.; Rossing, P.; Lambelet, M.; et al. Effect of finerenone on ambulatory blood pressure in chronic kidney disease in type 2 diabetes. J. Hypertens. 2023, 41, 295–302.
- Wojcik, C.; Warden, B.A. Mechanisms and Evidence for Heart Failure Benefits from SGLT2 Inhibitors. Curr. Cardiol. Rep. 2019, 21, 130.
- Gronda, E.; Vanoli, E.; Iacoviello, M.; Caldarola, P.; Gabrielli, D.; Tavazzi, L. The Benefit of Sodium-Glucose Co-Transporter Inhibition in Heart Failure: The Role of the Kidney. Int. J. Mol. Sci. 2022, 23, 11987.
- Bjornstad, P.; Greasley, P.J.; Wheeler, D.C.; Chertow, G.M.; Langkilde, A.M.; Heerspink, H.J.L.; Van Raalte, D.H. The Potential Roles of Osmotic and Nonosmotic Sodium Handling in Mediating the Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Heart Failure. J. Card. Fail. 2021, 27, 1447–1455.
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117.
- Zelniker, T.A.; Braunwald, E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 422–434.
- Varadhan, A.; Stephan, K.; Gupta, R.; Vyas, A.V.; Ranchal, P.; Aronow, W.S.; Hawwa, N.; Lanier, G.M. Growing role of SGLT2i in heart failure: Evidence from clinical trials. Expert. Rev. Clin. Pharmacol. 2022, 15, 147–159.
- Gupta, R.; Maitz, T.; Egeler, D.; Mehta, A.; Nyaeme, M.; Hajra, A.; Goel, A.; Sreenivasan, J.; Patel, N.; Aronow, W.S. SGLT2 inhibitors in hypertension: Role beyond diabetes and heart failure. Trends Cardiovasc. Med. 2022, in press.
- Kario, K.; Ferdinand, K.C.; O’Keefe, J.H. Control of 24-hour blood pressure with SGLT2 inhibitors to prevent cardiovascular disease. Prog. Cardiovasc. Dis. 2020, 63, 249–262.
- Kario, K.; Ferdinand, K.C.; Vongpatanasin, W. Are SGLT2 Inhibitors New Hypertension Drugs? Circulation 2021, 143, 1750–1753.
- Goldberg, L.R. The Pleiotropic Effects of SGLT2 Inhibitors: Remodeling the Treatment of Heart Failure. J. Am. Coll. Cardiol. 2021, 77, 256–258.
- Zannad, F.; Ferreira, J.P.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Ofstad, A.P.; Pfarr, E.; Jamal, W.; et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: A meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet 2020, 396, 819–829.
- Salazar, R.A.; Stroud, S.C.; DeFilippis, E.M. A Sweet Solution for Heart Failure with Preserved Ejection Fraction: The Role of Sodium-Glucose Cotransporter-2 Inhibitors. Circ. Heart Fail. 2023, 16, e010283.
- Li, M.; Yi, T.; Fan, F.; Qiu, L.; Wang, Z.; Weng, H.; Ma, W.; Zhang, Y.; Huo, Y. Effect of sodium-glucose cotransporter-2 inhibitors on blood pressure in patients with heart failure: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2022, 21, 139.
- Packer, M. How can sodium-glucose cotransporter 2 inhibitors stimulate erythrocytosis in patients who are iron-deficient? Implications for understanding iron homeostasis in heart failure. Eur. J. Heart Fail. 2022, 24, 2287–2296.
- Mancia, G.; Kjeldsen, S.E.; Kreutz, R.; Pathak, A.; Grassi, G.; Esler, M. Individualized Beta-Blocker Treatment for High Blood Pressure Dictated by Medical Comorbidities: Indications Beyond the 2018 European Society of Cardiology/European Society of Hypertension Guidelines. Hypertension 2022, 79, 1153–1166.
- Masarone, D.; Martucci, M.L.; Errigo, V.; Pacileo, G. The Use of beta-Blockers in Heart Failure with Reduced Ejection Fraction. J. Cardiovasc. Dev. Dis. 2021, 8, 101.
- Cleland, J.G.F.; Bunting, K.V.; Flather, M.D.; Altman, D.G.; Holmes, J.; Coats, A.J.S.; Manzano, L.; McMurray, J.J.V.; Ruschitzka, F.; van Veldhuisen, D.J.; et al. Beta-blockers for heart failure with reduced, mid-range, and preserved ejection fraction: An individual patient-level analysis of double-blind randomized trials. Eur. Heart J. 2018, 39, 26–35.
- Hogg, K.; McMurray, J. Neurohumoral pathways in heart failure with preserved systolic function. Prog. Cardiovasc. Dis. 2005, 47, 357–366.
- Meyer, M.; LeWinter, M.M. Heart Rate and Heart Failure with Preserved Ejection Fraction: Time to Slow beta-Blocker Use? Circ. Heart Fail. 2019, 12, e006213.
- Triposkiadis, F.; Xanthopoulos, A.; Starling, R.C. Medical Treatment of Heart Failure: Ignore the Ejection Fraction and Treat All? J. Card. Fail. 2021, 27, 907–909.
- Mullens, W.; Damman, K.; Harjola, V.P.; Mebazaa, A.; Brunner-La Rocca, H.P.; Martens, P.; Testani, J.M.; Tang, W.H.W.; Orso, F.; Rossignol, P.; et al. The use of diuretics in heart failure with congestion—A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 137–155.
- Wilcox, C.S.; Testani, J.M.; Pitt, B. Pathophysiology of Diuretic Resistance and Its Implications for the Management of Chronic Heart Failure. Hypertension 2020, 76, 1045–1054.
- Shah, S.U.; Anjum, S.; Littler, W.A. Use of diuretics in cardiovascular diseases: (1) heart failure. Postgrad. Med. J. 2004, 80, 201–205.
- Weir, M.R.; Zappe, D.; Orloski, L.A.; Sowers, J.R. How early should blood pressure control be achieved for optimal cardiovascular outcomes? J. Hum. Hypertens. 2011, 25, 211–217.
- Agarwal, R.; Joseph, A.; Anker, S.D.; Filippatos, G.; Rossing, P.; Ruilope, L.M.; Pitt, B.; Kolkhof, P.; Scott, C.; Lawatscheck, R.; et al. Hyperkalemia Risk with Finerenone: Results from the FIDELIO-DKD Trial. J. Am. Soc. Nephrol. 2022, 33, 225–237.
- Paczkowska-Walendowska, M.; Sip, S.; Staszewski, R.; Cielecka-Piontek, J. Single-Pill Combination to Improve Hypertension Treatment: Pharmaceutical Industry Development. Int. J. Environ. Res. Public. Health 2022, 19, 4156.
- Perrone, V.; Veronesi, C.; Gambera, M.; Nati, G.; Perone, F.; Tagliabue, P.F.; Degli Esposti, L.; Volpe, M. Treatment with Free Triple Combination Therapy of Atorvastatin, Perindopril, Amlodipine in Hypertensive Patients: A Real-World Population Study in Italy. High. Blood Press. Cardiovasc. Prev. 2019, 26, 399–404.
- Gaciong, Z. Preference and Adherence to a Fixed-Dose Combination of Bisoprolol-Aspirin and Blood Pressure Control: Results of an Open-Label, Multicentre Study. J. Clin. Med. 2022, 12, 17.
- Taylor, J.L.; Myers, J.; Bonikowske, A.R. Practical guidelines for exercise prescription in patients with chronic heart failure. Heart Fail. Rev. 2023.
- Crisci, G.; De Luca, M.; D’Assante, R.; Ranieri, B.; D’Agostino, A.; Valente, V.; Giardino, F.; Capone, V.; Chianese, S.; Rega, S.; et al. Effects of Exercise on Heart Failure with Preserved Ejection Fraction: An Updated Review of Literature. J. Cardiovasc. Dev. Dis. 2022, 9, 241.
- Olsen, L.N.; Fischer, M.; Evans, P.A.; Gliemann, L.; Hellsten, Y. Does Exercise Influence the Susceptibility to Arterial Thrombosis? An Integrative Perspective. Front. Physiol. 2021, 12, 636027.
- Daniela, M.; Catalina, L.; Ilie, O.; Paula, M.; Daniel-Andrei, I.; Ioana, B. Effects of Exercise Training on the Autonomic Nervous System with a Focus on Anti-Inflammatory and Antioxidants Effects. Antioxidants 2022, 11, 350.
- Boulmpou, A.; Theodorakopoulou, M.P.; Boutou, A.K.; Alexandrou, M.E.; Papadopoulos, C.E.; Bakaloudi, D.R.; Pella, E.; Sarafidis, P.; Vassilikos, V. Effects of different exercise programs on the cardiorespiratory reserve in HFpEF patients: A systematic review and meta-analysis. Hell. J. Cardiol. 2022, 64, 58–66.
- Lopes, S.; Afreixo, V.; Teixeira, M.; Garcia, C.; Leitao, C.; Gouveia, M.; Figueiredo, D.; Alves, A.J.; Polonia, J.; Oliveira, J.; et al. Exercise training reduces arterial stiffness in adults with hypertension: A systematic review and meta-analysis. J. Hypertens. 2021, 39, 214–222.
- Rahman, M.M.; Islam, F.; Or-Rashid, M.H.; Mamun, A.A.; Rahaman, M.S.; Islam, M.M.; Meem, A.F.K.; Sutradhar, P.R.; Mitra, S.; Mimi, A.A.; et al. The Gut Microbiota (Microbiome) in Cardiovascular Disease and Its Therapeutic Regulation. Front. Cell Infect. Microbiol. 2022, 12, 903570.
- Yuan, L.; Li, Y.; Chen, M.; Xue, L.; Wang, J.; Ding, Y.; Gu, Q.; Zhang, J.; Yang, R.; Zhao, H.; et al. Effects of probiotics on hypertension. Appl. Microbiol. Biotechnol. 2023, 107, 1107–1117.
- Zarezadeh, M.; Musazadeh, V.; Ghalichi, F.; Kavyani, Z.; Nasernia, R.; Parang, M.; Jamilian, P.; Jamilian, P.; Fakhr, L.; Ostadrahimi, A.; et al. Effects of probiotics supplementation on blood pressure: An umbrella meta-analysis of randomized controlled trials. Nutr. Metab. Cardiovasc. Dis. 2023, 33, 275–286.
- Nagai, M.; Dote, K.; Forster, C.Y. Denervation or stimulation? Role of sympatho-vagal imbalance in HFpEF with hypertension. Hypertens. Res. 2023, 46, 1727–1737.
- Barbato, E.; Azizi, M.; Schmieder, R.E.; Lauder, L.; Bohm, M.; Brouwers, S.; Bruno, R.M.; Dudek, D.; Kahan, T.; Kandzari, D.E.; et al. Renal denervation in the management of hypertension in adults. A clinical consensus statement of the ESC Council on Hypertension and the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur. Heart J. 2023, 44, 1313–1330.
- Pavlov, V.A.; Tracey, K.J. The vagus nerve and the inflammatory reflex—Linking immunity and metabolism. Nat. Rev. Endocrinol. 2012, 8, 743–754.
- Stavrakis, S.; Elkholey, K.; Morris, L.; Niewiadomska, M.; Asad, Z.U.A.; Humphrey, M.B. Neuromodulation of Inflammation to Treat Heart Failure with Preserved Ejection Fraction: A Pilot Randomized Clinical Trial. J. Am. Heart Assoc. 2022, 11, e023582.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
836
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
2 times
(View History)
Update Date:
09 Nov 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No