Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ane Larrea | -- | 3616 | 2023-11-02 09:46:55 | | | |
| 2 | Rita Xu | Meta information modification | 3616 | 2023-11-02 09:59:03 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Larrea, A.; Elexpe, A.; Díez-Martín, E.; Torrecilla, M.; Astigarraga, E.; Barreda-Gómez, G. Neuroinflammation in Stroke and Trauma Patients. Encyclopedia. Available online: https://encyclopedia.pub/entry/51077 (accessed on 28 July 2026).
Larrea A, Elexpe A, Díez-Martín E, Torrecilla M, Astigarraga E, Barreda-Gómez G. Neuroinflammation in Stroke and Trauma Patients. Encyclopedia. Available at: https://encyclopedia.pub/entry/51077. Accessed July 28, 2026.
Larrea, Ane, Ane Elexpe, Eguzkiñe Díez-Martín, María Torrecilla, Egoitz Astigarraga, Gabriel Barreda-Gómez. "Neuroinflammation in Stroke and Trauma Patients" Encyclopedia, https://encyclopedia.pub/entry/51077 (accessed July 28, 2026).
Larrea, A., Elexpe, A., Díez-Martín, E., Torrecilla, M., Astigarraga, E., & Barreda-Gómez, G. (2023, November 02). Neuroinflammation in Stroke and Trauma Patients. In Encyclopedia. https://encyclopedia.pub/entry/51077
Larrea, Ane, et al. "Neuroinflammation in Stroke and Trauma Patients." Encyclopedia. Web. 02 November, 2023.
Copy Citation
Neuroinflammation has a significant impact on different pathologies, such as stroke or spinal cord injury, intervening in their pathophysiology: expansion, progression, and resolution. Neuroinflammation involves oxidative stress, damage, and cell death, playing an important role in neuroplasticity and motor dysfunction by affecting the neuronal connection responsible for motor control.
neuroinflammation
stroke
traumatic injury
1. Introduction
Neuroinflammation is a highly complex process characterized by the activation of various glial cells and the release of proinflammatory mediators in the central nervous system (CNS) [1]. It develops secondary to a variety of stimuli, including traumatic injuries, infections, and neurodegenerative processes [2]. Thus, neuroinflammation has emerged as a key research focus, given its significant impact on neuronal function and neurological and motor recovery from different conditions due to its close relationship with modulation of cellular responses and neural homeostasis [3].
Spinal cord injury (SCI) involves the physical and/or functional disruption of neuronal connections in the spinal cord, affecting the integrity of electrical and chemical signals necessary for proper motor, sensory, and autonomic function [3]. This anatomical damage also triggers an inflammatory response in the CNS. Activation of microglia and astrocytes initiates a molecular signaling cascade involving the release of proinflammatory cytokines and reactive oxygen species (ROS) [2]. This exacerbated inflammation perpetuates neuronal damage and promotes scarring and fibrous tissue formation, resulting in chronic and persistent disability [4]. Cerebral ischemia also activates glial cells in response to tissue stress, resulting in the release of cytokines and chemokines that amplify the inflammatory response [5]. Like SCI, strokes also result in neuroinflammation as an integral component of pathogenesis due to the sudden interruption of blood flow to a part of the brain, which can result in the sudden loss of cognitive, sensory, and/or motor functions [6]. As brain cells die from a lack of oxygen and nutrients, the release of proinflammatory factors is further increased, exacerbating tissue damage and hindering functional recovery [7].
The multidisciplinary approach to addressing neuroinflammation in the context of SCI and stroke is significantly enhanced by state-of-the-art diagnostic techniques. Neuroimaging techniques have emerged as fundamental tools for the accurate assessment of neuroinflammation. Magnetic resonance imaging (MRI) [8], computed tomography (CT) [9], positron emission tomography (PET) [10], and contrast-enhanced ultrasound (CEUS) [11] offer specific visualization of lesions, providing information about their location, extent, and relationship to surrounding structures. This ability to accurately map damage and identify areas affected by inflammation translates into a more comprehensive understanding of the disease, which in turn guides therapeutic decisions and provides a solid basis for prognosis [12]. On the other hand, biomarkers are highly informative molecules that are released into the bloodstream in response to neuroinflammation [13]. These biological indicators, whose presence and concentration can be detected with specific methods, are emerging as promising diagnostic tools [14]. Their ability to provide a window into the internal state of the CNS, even in the absence of overt clinical manifestations, provides potential for early diagnosis and monitoring of disease progression [15]. By analyzing these biomarkers, detailed information is obtained about the degree of inflammation present, immune system response, and cellular activity in the compromised neural tissue. This information can not only accelerate the diagnostic stage but also allow for continuous and adaptive monitoring of the disease course, which is essential for the development of more effective and personalized therapeutic strategies [14][15][16].
In the therapeutic field, the incorporation of electrostimulation and bionanomaterials has arisen as cutting-edge strategies. Electrostimulation has emerged as a highly promising technique to precisely modulate neuronal activity and mitigate the inflammatory responses characteristically observed in SCI and stroke [17]. This therapeutic modality leverages the fundamental principles of bioelectronics and neuroscience to influence the electrical behavior of nerve cells and glial cells, with results in terms of improved motor function, recovery of mobility, and reduction in inflammation in affected regions [18]. Bionanomaterials have proven to be an innovative option in the search for effective strategies to address the implications of neuroinflammation in SCI and stroke [19][20]. These materials are designed to interact at the nanometer scale with biological tissues. In addition, they exhibit unique characteristics that allow them to act in versatile and highly specific ways for the controlled delivery of therapeutic agents [21]. This nanotechnological approach has enabled the encapsulation and gradual release of anti-inflammatory molecules and growth factors in areas affected by neuroinflammation, which in turn promotes neuronal regeneration, angiogenesis, and reduction in the local inflammatory response [21]. This approach, targeted to the site of injury or the ischemic area in the case of stroke, has great potential to mitigate the adverse effects of neuroinflammation and promote functional recovery and, consequently, motor function [22]. Both strategies converge in their goal of restoring balance and promoting repair in the affected nerve tissue. The combination of these innovative therapeutic strategies, with the constantly evolving research on the mechanisms of neuroinflammation, offers a comprehensive approach to improving patients’ recovery and quality of life.
2. Neuroinflammation
Neuroinflammation includes several pathological processes, ranging from altered morphology of glial cells to invasion and destruction of tissues by immune cells migrating from the periphery [23][24][25][26]. The immune system maintains a close relationship with the nervous system, as central nervous system (CNS) cells can be activated by peripheral inflammatory mediators, and peripheral immune cells can infiltrate into the brain [25]. In fact, chronic neuroinflammation can alter learning, cognitive, and motor functions by altering neurotransmission [27], becoming an important risk factor for the development of neuropsychological diseases such as Schizophrenia, Bipolar Disorder [28], Mayor Depressive Disorder [29][30][31], or Parkinson [32].
Microglia cells are the main recipients of peripheral inflammatory signals reaching the brain. Once activated, an inflammatory cascade is initiated with the release of chemokines, cytokines, and reactive oxygen and nitrogen species (ROS and RNS, respectively), triggering the activation of astrocytes and, thus, amplifying the inflammatory signal within the CNS. Several astrocyte functions will be altered, resulting in the dysregulation of neurotrophic factor production, transporter function, and neurotransmitter synthesis. The toxic effects of overexposure to cytokines also affect oligodendrocytes, with subsequent apoptosis and demyelination of neurons. Thus, excessive release of proinflammatory mediators together with incorrect neurotransmitter reuptake, decreased release of neurotrophic factors, and oxidative stress cause damage to neuronal plasticity, leading to neurodegeneration and apoptosis [24] (Figure 1).
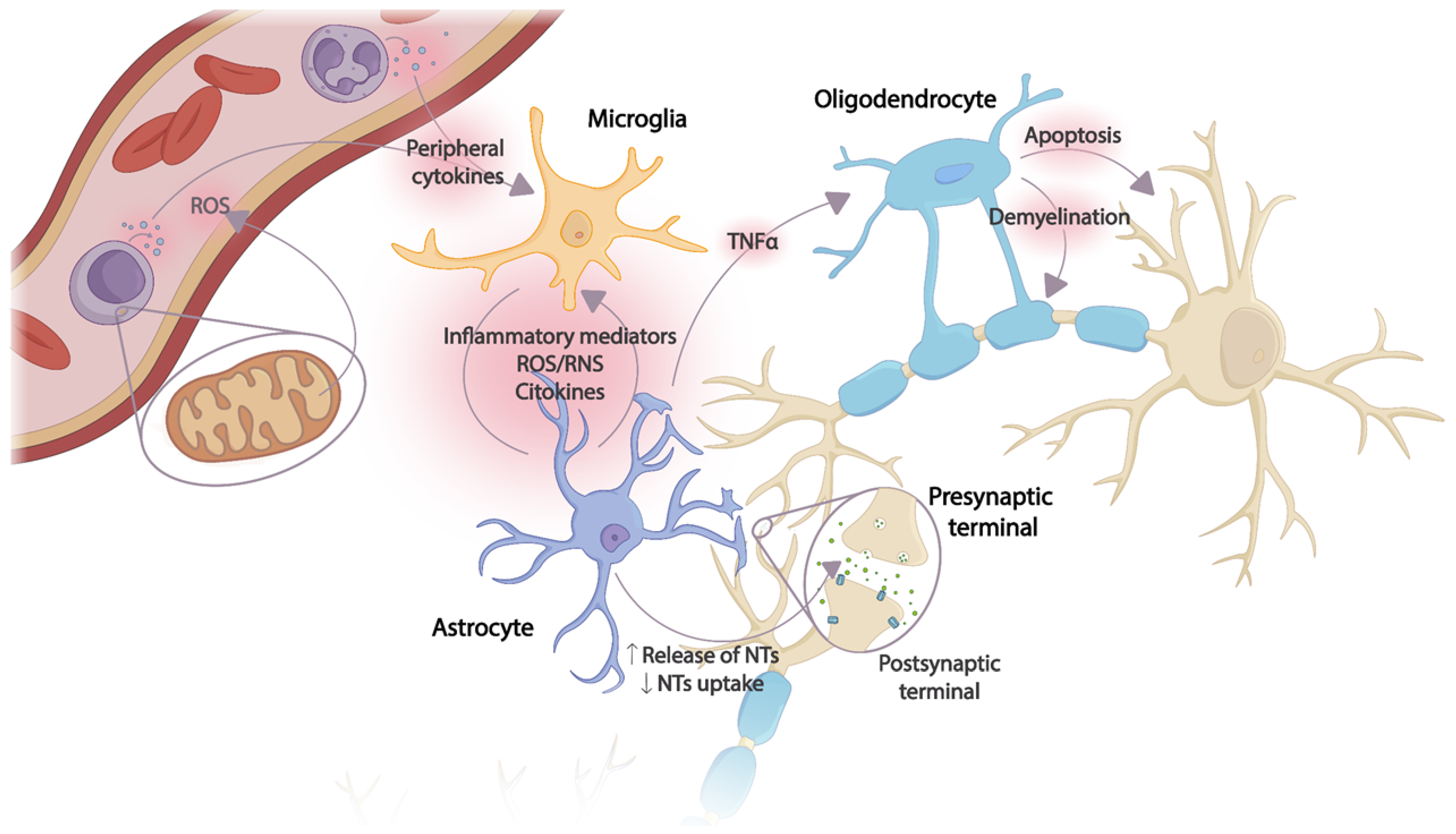
Figure 1. Effects of the CNS inflammatory cascade on neuronal plasticity caused by an uncontrolled peripheral inflammatory response. The production of peripheral proinflammatory mediators originating from ROS and mitochondrial dysfunction in immune cells activates microglia. An inflammatory cascade is triggered in which the release of cytokines and other inflammatory mediators induces astrocyte activation, thus amplifying the inflammatory signal in the CNS. Several astrocyte functions are altered due to continuous exposure to cytokines, inflammatory mediators, and ROS/RNS. Oligodendrocytes, especially sensitive to the toxic effect of TNF-α, induce apoptosis and demyelination. NTs: Neurotransmitters; ROS: reactive oxygen species; RNS: reactive nitrogen species; TNF-α: tumor necrosis factor.
2.1. Stroke
Strokes are those disorders that produce functional and structural neuronal alterations in different areas of the brain due to maintained hypoxia, a consequence of an abrupt variation (interruption or reduction) in cerebral circulation in such regions [33]. Thus, stroke causes transitory or definitive deficits in their functioning, causing sensory, motor, and cognitive alterations [34].
Strokes can be classified into two subtypes based on the cause that determines the presence of the pathology:
- -
-
Ischemic stroke (80%): Occurs because of a decrease and insufficiency of blood supply to the CNS, causing a circumscribed area of cerebral infarction. Depending on their etiology, strokes can be subclassified as thrombotic due to the formation of a blood clot in an area of the brain and embolic because of the formation of a blood clot in another cerebral artery that subsequently travels to the brain. When the symptoms last less than 24 h, it is called a transient ischemic attack (TIA [35][36]).
- -
-
Hemorrhagic stroke (20%): Is due to parenchymal and/or subarachnoid bleeding. Generally, they are caused by arterial hypertension (AHT), aneurysm ruptures, and or arteriovenous malformations [37].
Depending on the affected area, the altered functions will vary, although it is very common that the stroke involves the pyramidal system, causing a first motor neuron or upper motor neuron syndrome [35]. The clinical manifestations can be classified depending on whether they refer to a loss or decrease in functions (negative clinical manifestations) or the appearance of new or abnormal functions (positive clinical manifestations) [37][38]. On the one hand, the negative clinical manifestations include abolition of superficial reflexes, as well as paralysis or paralytic of muscles. On the other hand, the positive clinical manifestations include muscle spasticity (in antigravitational musculature) and hyperreflexia of the musculature, whose centers are in the intralesional area, appearing pathological reflexes and clonus [37].
Neuroinflammation and Stroke
Following ischemia, a neuropathological cascade of mechanisms is activated that triggers innate and potentially adaptive inflammatory and immune responses in the central and peripheral nervous systems. This activation leads to the extension and deterioration of the brain injury [6].
The progression and increase in neuroinflammation are directly linked to the immune system, as reflected by the increase in damage-associated molecular patterns (DAMPs) to nuclear or cytosolic proteins observed after stroke [39]. Thus, cells of the innate immune system, such as neutrophils, macrophages/microglia, and astrocytes, are activated [12]. In turn, there is also activation of T cells, regulatory T cells, and B cells of the adaptive immune system, which are able to specifically recognize antigens presented in the context of major histocompatibility complex molecules on antigen-presenting cells [40]. In CNS, infiltrating T cells are mainly CD4+ T cells (helper) [41] and CD8+ T cells (cytotoxic) [42].
Importantly, in the acute phase, immediately after stroke, neuroinflammation may play an endogenous neuroprotective role by phagocytizing leukocytes brain cells and increasing immune cell signaling [43]. This action increases the expression of anti-inflammatory cytokines that facilitate axonal recovery and repair [39]. T-helper cells may have a dual role in neuroinflammation, as, on the one hand, they can secrete anti-inflammatory cytokines that can limit the inflammatory response and protect brain tissue [40]. However, on the other hand, they trigger the release of potent proinflammatory cytokines into cerebrospinal fluid and blood, increasing infarction and cell apoptosis (Figure 2) [39][40][43].
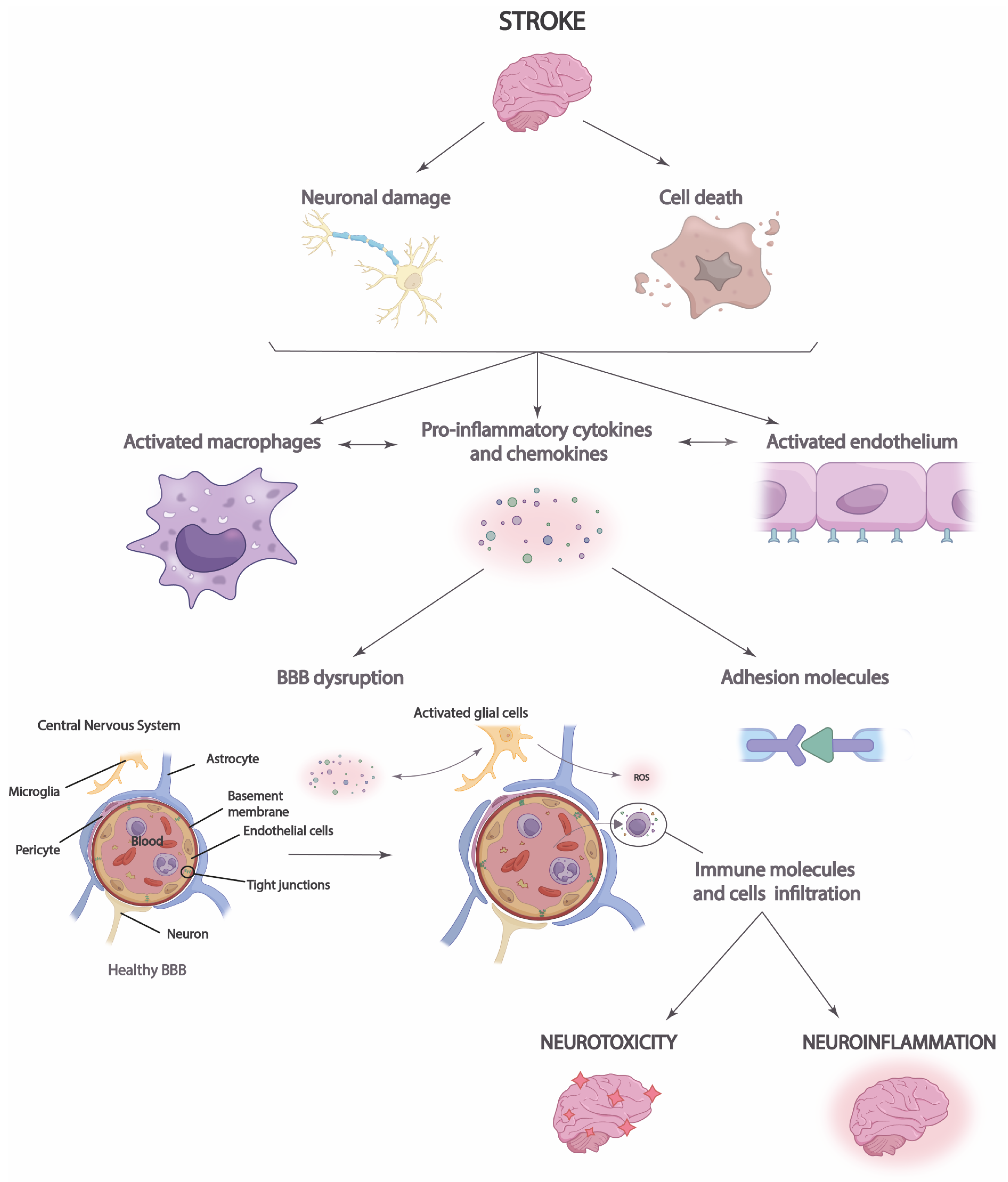
Figure 2. Neuroinflammation process in stroke. After a stroke, cell damage and neuronal death occur, triggering the increased release of chemokines and proinflammatory cytokines that lead to blood–brain barrier (BBB) disruption and immune cell infiltration. This causes brain neurotoxicity and neuroinflammation.
Moreover, it is necessary to highlight the role that neuroinflammation plays in the integrity of the blood–brain barrier (BBB) and vice versa [44]. Disruption of the BBB allows immune cells, inflammatory molecules, and serum proteins to penetrate the brain parenchyma from the periphery [45]. This causes the migration of prostaglandins, proinflammatory cytokines, and other mediators to the site of injury, which increases the number of immune cells and microglia [44]. Thus, the inflammatory response and brain damage are aggravated in the ischemic penumbra, the region surrounding the infarct area at high risk of further damage [46]. Neuroinflammation in the ischemic penumbra can be particularly detrimental, as it can contribute to cell death in this area, which enlarges the size of the cerebral infarct and aggravates the clinical consequences of stroke. Disruption of the BBB may also have long-term consequences after stroke [12]. The influx of immune cells and inflammatory molecules can perpetuate neuroinflammation, which may contribute to the progression of brain damage and scar formation in the affected tissue [45]. In addition, BBB dysfunction may affect the regulation of cerebral blood flow and homeostasis of the brain environment, which may influence functional recovery and brain plasticity after stroke [44]. Likewise, neuronal antigen response may be induced, and chronic cell death may be increased, perpetuating long-term neuroinflammation [12][44][45].
2.2. Spinal Cord Injury
SCI is a pathological process of any etiology that affects the spinal cord and causes transitory or permanent impairment of motor, sensory, and autonomic function [3]. The annual incidence of SCI is approximately 11.4 to 53.4 per million population worldwide, and its etiology may be due to traumatic (80%) or non-traumatic causes (congenital or secondary to disease). SCI can be classified according to [47]:
- -
-
Cause: Traumatic or non-traumatic.
- -
-
Mechanism of injury: Hyperflexion, flexion with rotation, hyperextension, or compression.
- -
-
Level of injury: Cervical, dorsal, or/and lumbosacral.
- -
-
Extension: Complete or incomplete.
The assessment of motor and sensory functions is performed according to international standards via the American Spinal Injury Association (ASIA) Impairment Scale [47]. The prognostic factor is determined by the evaluation of the ASIA scale 72 h after the injury, with the maximum risk of mortality in the first year [47].
It is important to know the extension of the SCI since incomplete SCI causes specific syndromes: Scheiner´s syndrome, anterior spinal artery syndrome [48], spinal cord hemisection [49], posterior cord [50], and cauda equina syndrome [51]. All of these syndromes preserve some spinal cord function below the level of the lesion. However, in the case of complete SCI, all functions below the lesion are abolished [52].
SCI shows different clinical phases. The first is the spinal shock phase, immediately after the injury, which extends up to the second and eighth weeks [53][54]. This phase is identified as the most severe since motor, sensory, and vegetative functions of the lesional and infralateral segments are interrupted [53]. At this time, motor disturbances are characteristic of the lower motor neuron [55]. After the spinal shock, the phase of spinal automatism appears, where the spinal reflex center and activities in the intralesional segment are recovered [56] (except in cauda equina lesions [51]), and even the alteration of the injured segment persists. In this case, the typical motor alteration is of the upper motor neuron [56].
Neuroinflammation and Spinal Cord Injury
Following SCI, a range of vascular, cellular, and molecular alterations originate in the CNS and produce imbalances between immune cells and modulatory factors resulting from neuroinflammation secondary to trauma [57]. Although these may have a dual effect in helping to regulate axonal homeostasis and healing, the imbalance in production results in increased axonal and tissue damage and cell death, aggravating the initial situation, course, and prognosis of SCI [2][58].
Acute neuroinflammation develops in several stages (Figure 3). In the first stage, the release of proinflammatory cytokines, chemokines, and ROS by microglia, astrocytes, and peripheral immune cells is induced [58]. In this way, a cascade activation of inflammatory and immune pathways is caused, attracting the presence of a greater number of immune cells to the site of the lesion [59]. In the second stage, macrophage and T-cell infiltration occurs [60], increasing pro-inflammatory cytokine and pro-inflammatory chemokine proliferation. Finally, in the third stage, BBB injury occurs, resulting in the migration of leukocytes to the area of the lesion [61].
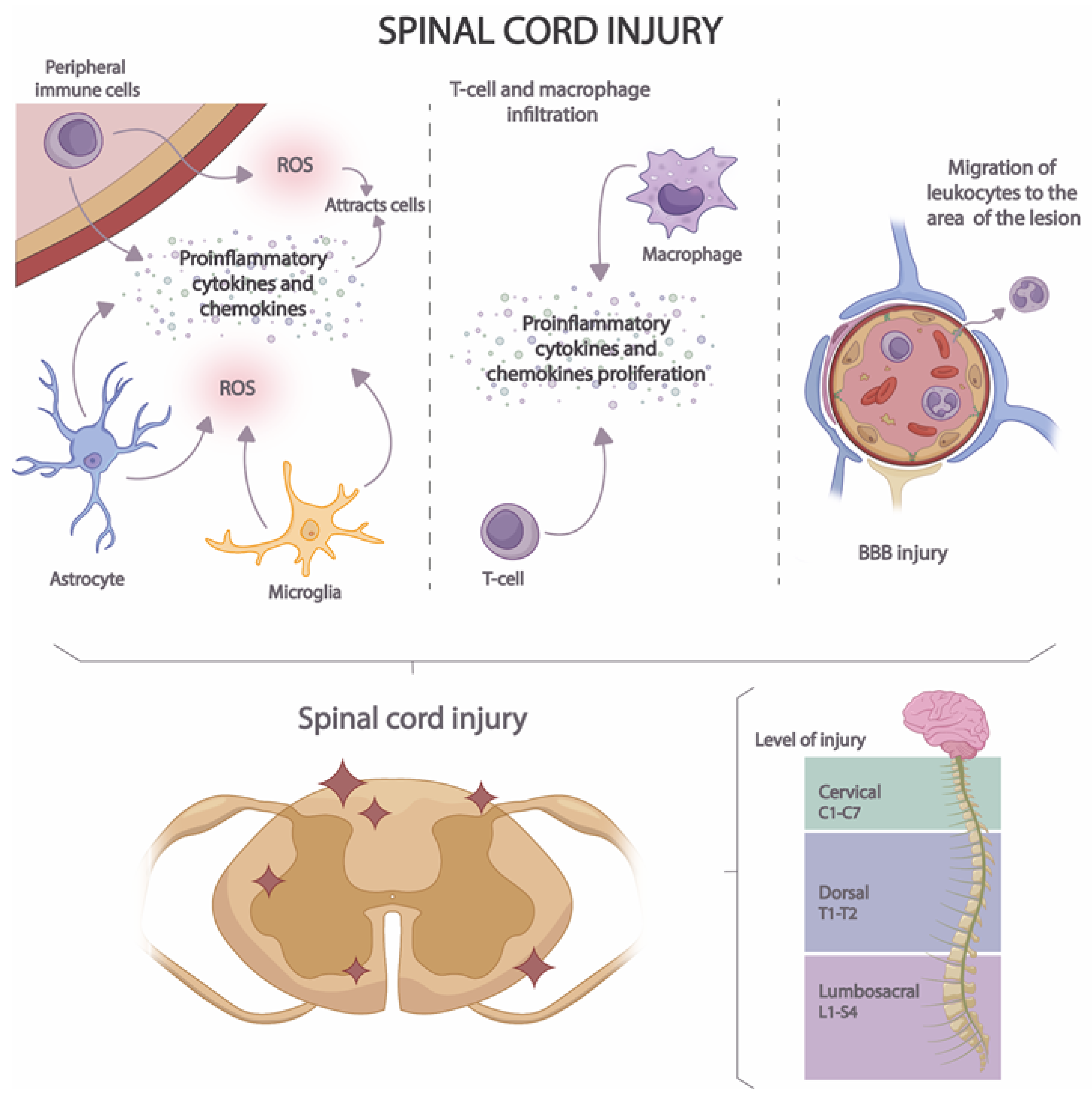
Figure 3. Stages of neuroinflammation in SCI.
BBB is a highly specialized structure that separates the peripheral blood from the CNS and protects the brain from the entry of potentially harmful substances and immune cells [62]. The BBB is composed mainly of endothelial cells with tight junctions that form a highly selective barrier to the passage of molecules [62][63]. It is also surrounded by glial cells that contribute to maintaining the integrity of the barrier [64]. BBB injury results in increased permeability and migration of immune molecules and cells into neuronal tissue [65]. The injury is produced by the activation of microglia and astrocytes and the release of proinflammatory cytokines and chemokines that induce the expression of adhesion molecules on endothelial cells [2]. These cells are also damaged by increased production of ROS and by the action of proteolytic enzymes that lead to the disruption of endothelial cell junctions [63]. Moreover, the expression of endothelial cell transporters and proteins is also modified during neuroinflammation [66], thus altering the regulation of the flow of molecules. All these processes lead to the amplification of the inflammatory response.
The close relationship between neuroplasticity and neuroinflammation in the context of SCI should be emphasized. The presence of edema and an increase in BBB permeability, as well as the release of proinflammatory cytokines, chemokines, and ROS, affect neuronal reorganization in the area of the lesion [67]. Likewise, after SCI, the unaffected areas also undergo changes in connections and functions in an attempt to compensate for the functional loss secondary to the injury [68]. Consequently, neuronal circuits, synaptic plasticity, and activation of non-injured motor areas are reorganized to compensate for lost functions [69]. The neuroplasticity of uninjured areas can be affected by neuroinflammation since inflammatory mediators can positively alter synaptic signaling and, thus, neuronal plasticity [69][70]. Additionally, neurotrophic mediators affect the survival and growth of neurons after SCI growth of neurons after SCI, attenuating part of the deleterious effects triggered by mitochondrial dysfunction and oxidative stress [71].
2.3. Neuroinflammation and Mitochondrial Activity
Mitochondria are recognized as powerhouses, present in virtually all eukaryotic cells. They are dynamic organelles that constantly fuse and divide to regulate their shape, size, number, and bioenergetic function [72]. In fact, there is a variable number of mitochondria in the cellular medium, and their number is directly related to the energy needs of the cell [73][74]. They are responsible for carrying out several functions, such as calcium homeostasis [75], programmed cell death or apoptosis [76], synaptic plasticity, adenosine triphosphate (ATP) synthesis via the tricarboxylic acid cycle (TAC), and OXPHOS and ROS production and elimination [77][78]. ROS are chemical compounds that are formed after incomplete reduction in oxygen [79]. They are natural metabolites generated in normal cellular activity that participate in cell signaling. However, an imbalance between ROS production and the antioxidant defense system in the organism leads to disruption of cellular function and toxicity. This can occur due to an overproduction of ROS or a decrease in the antioxidant defense mechanism [80].
In this sense, oxidative stress derived from the increase in the ROS production at the neuronal level and in cells of the peripheral system causes a decrease in the generation of ATP that will eventually lead to a lack of energy at times of increased energy demand, for example, in neuronal activity to modulate synaptic connections and neuronal plasticity [77] or under conditions of stress and inflammation [81], factors that have often been related to different neurodegenerative diseases [82][83]. Inflammation is a physiological response of the immune system that promotes the mobilization of immune cells to the site of infection or damage to eliminate the triggering factor, repair the damaged tissue, and restore the homeostasis of the organism. Cellular energy metabolism is an important part of the machinery that ensures the proper functioning of immune [1]. Without adequate energy, immune function would fail, altering immune responses or triggering uncontrolled activation [81]. This process would end up damaging and fragmenting mitochondrial DNA that will be released first to the cytosol and then to the extracellular medium by various mechanisms, including transport in mitochondria-derived vesicles (MVD) or via mitochondrial permeability transition pores (MPT). This mitochondrial DNA (mtDNA) acts as a potent DAMP (damage-associated molecular patterns), activating the TLR9-mediated signaling pathway that will ultimately lead to increased production of proinflammatory mediators, such as TNF and IL-6 [84]. Taken together, inflammation can impair mitochondrial function, while alterations in mitochondrial activity may promote uncontrolled inflammatory responses, creating a vicious cycle that can ultimately compromise neuronal function at the bioenergetic level [85].
2.4. Cytokines and Chemokines Involved in Neuroinflammation
Cytokines and chemokines are cell signaling molecules that play a crucial role in regulating the immune response and communication between different cell types in the body. However, in the context of SCI and stroke, the interaction of these molecules can have both beneficial and detrimental effects on neuroinflammation (Table 1).
Table 1. Cytokines and chemokines involved in neuroinflammation: Detrimental and beneficial effects related to each molecule.
| Cytokines and Chemokines | Detrimental Effects | Beneficial Effects | Secretory Cell | Refs. |
|---|---|---|---|---|
| Interleukin 1β (IL-1β) | Increased secondary brain damage Increased BBB permeability |
Tissue recovery Apoptosis inhibition |
Microglia Macrophages |
[86] |
| Interleukin 1α (IL-1α) | Chronic inflammation Damage of tissue Autoimmune pathologies |
Tissue recovery Activation immune system |
Microglia | [2][18] |
| Interleukin 1F1 (IL-1F1) | Increased inflammatory response, hypersensitivity, and autoimmune diseases | Regulation of the immune response Tissue recovery. Neuroprotective function. |
Neutrophils | [87] |
| Interleukin 1F2 (IL-1F2) | Increased prostaglandins, cyclooxygenase 2, and phospholipase A2 | Regulation of the immune response. Tissue recovery |
Dendritic cells, macrophages, endothelial, and T cells | [87] |
| Interleukin 12 (IL-12) | Increased immune response Difficulty axonal regeneration |
Activation immune system Elimination death cells |
Dendritic cells, macrophages, monocytes, neutrophils, microglia, and T-cells. | [88] |
| Interleukin 17 (IL-17) | Damage of BBB Increased immune response |
Antipathogenic response Decontrol immune cells |
T helper, dendritic cells, and macrophages | [89][90] |
| Tumor Necrosis Factor α (TNF-α) | Neurotoxicity Increased BBB permeability |
Tissue recovery Antipathogenic response |
Microglia, neurons, astrocytes, monocytes, and oligodendrocytes | [2][18][91] |
| Interferon γ (IFN-γ) | Neurotoxicity Difficulty neuroplasticity |
Antipathogenic response Tissue recovery |
γδ T-cells | [2] |
| Interleukin 5 (IL-5) | Allergic response Decreased immune response |
Regulation of allergic pathologies Antipathogenic response |
Hematopoietic and non-hematopoietic cells, granulocytes, T, and natural helper cells | [88][92] |
| Interleukin 10 (IL-10) | Neurotoxicity Increased inflammatory response |
Inhibition TNF-α; IL-1; IL-6 Limitation inflammatory response |
T and B cells, monocytes, dendritic, and natural killer cells | [92] |
| Interleukin 4 (IL-4) | Immunosuppression | Inhibition TNF-α; IL-1; IL-6 Limitation inflammatory response |
T helper cells, eosinophils, and eosinophils | [93][94] |
| Interleukin 6 (IL-6) | Neurotoxicity Increased inflammatory response |
Antipathogenic response Increased axonal regeneration |
Astrocytes, microglia, and neurons | [95][96] |
| Interleukin 8 (IL-8) | Chronic inflammation Cardiovascular and pulmonary diseases |
Tissue recovery Neutrophills quimiotaxis |
Monocytes, endothelial cells, macrophages, and T cells. | [15] |
| C-C Motif Chemokine Ligand 2 (CCL 2) | Chronic inflammation Autoimmune diseases Increased cancer cell migration |
Regulation of immune response Angiogenesis Monocyte chemoattraction |
Activated T cells, astrocytes, microglia, and monocytes | [2][97] |
| C-C Motif Chemokine Ligand 3 (CCL 3) | Increased production of proinflammatory cytokines | Regulation of inflammatory response | Monocytes, macrophages, and dendritic cells | [98][99][100] |
| C-C Motif Chemokine Ligand 5 (CCL 5) | Chronic inflammation Cardiovascular diseases Neurological disorders |
Immune cells quimiotaxis Regulation of immune response Antiviral response |
IL-1 and macrophage migration inhibitory factor | [98][101] |
In certain situations, cytokines and chemokines can be beneficial in the response to SCI and stroke. They can recruit immune system cells and migrate to the site, which is essential to eliminate damaged tissues and toxic substances, as well as to initiate repair. However, overexpression or dysregulation of certain cytokines and chemokines can have detrimental effects. Excessive cytokine release causes excessive attraction of inflammatory cells to the site of injury and can lead to the formation of a toxic environment and excessive scarring that hinders neuronal regeneration. It also contributes to a chronic inflammatory environment. Thus, if the inflammatory response persists in an uncontrolled manner, it can contribute to secondary neuronal death and worsening damage.
Thus, cytokines and chemokines are molecules with a significant influence on neuroinflammation. Their role is complex and depends on the amount and type of molecules released, as well as their interaction with the cellular environment. Therefore, the balance between cytokines and chemokines is crucial in neuroinflammation associated with SCI and stroke.
References
- Breda, C.N.d.S.; Davanzo, G.G.; Basso, P.J.; Saraiva Câmara, N.O.; Moraes-Vieira, P.M.M. Mitochondria as Central Hub of the Immune System. Redox Biol. 2019, 26, 101255.
- Lukacova, N.; Kisucka, A.; Bimbova, K.K.; Bacova, M.; Ileninova, M.; Kuruc, T.; Galik, J. Glial-Neuronal Interactions in Pathogenesis and Treatment of Spinal Cord Injury. Int. J. Mol. Sci. 2021, 22, 13577.
- Silva, N.A.; Sousa, N.; Reis, R.L.; Salgado, A.J. From Basics to Clinical: A Comprehensive Review on Spinal Cord Injury. Prog. Neurobiol. 2014, 114, 25–57.
- Anderson, M.A.; Ao, Y.; Sofroniew, M.V. Heterogeneity of Reactive Astrocytes. Neurosci. Lett. 2014, 565, 23–29.
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The Science of Stroke: Mechanisms in Search of Treatments. Neuron 2010, 67, 181–198.
- Kamel, H.; Iadecola, C. Brain-Immune Interactions and Ischemic Stroke: Clinical Implications. Arch. Neurol. 2012, 69, 576–581.
- Anthony, S.; Cabantan, D.; Monsour, M.; Borlongan, C.V. Neuroinflammation, Stem Cells, and Stroke. Stroke 2022, 53, 1460–1472.
- Sillerud, L.O.; Yang, Y.; Yang, L.Y.; Duval, K.B.; Thompson, J.; Yang, Y. Longitudinal Monitoring of Microglial/Macrophage Activation in Ischemic Rat Brain Using Iba-1-Specific Nanoparticle-Enhanced Magnetic Resonance Imaging. J. Cereb. Blood Flow. Metab. 2020, 40, S117–S133.
- Campbell, B.C.V. Advances in Stroke Medicine. Med. J. Aust. 2019, 210, 367–374.
- Campbell, B.C.V.; Mitchell, P.J.; Kleinig, T.J.; Dewey, H.M.; Churilov, L.; Yassi, N.; Yan, B.; Dowling, R.J.; Parsons, M.W.; Oxley, T.J.; et al. Endovascular Therapy for Ischemic Stroke with Perfusion-Imaging Selection. N. Engl. J. Med. 2015, 372, 1009–1018.
- Kunte, H.; Schmidt, C.; Harms, L.; Rückert, R.I.; Grigoryev, M.; Fischer, T. Contrast-Enhanced Ultrasound and Detection of Carotid Plaque Neovascularization. Neurology 2012, 79, 2081.
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486.
- Sonawane, M.D.; Nimse, S.B. C-Reactive Protein: A Major Inflammatory Biomarker. Anal. Methods 2017, 9, 3400–3413.
- Strimbu, K.; Tavel, J.A. What Are Biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466.
- Simats, A.; García-Berrocoso, T.; Montaner, J. Neuroinflammatory Biomarkers: From Stroke Diagnosis and Prognosis to Therapy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1862, 411–424.
- de la Fuente, M.; Rodríguez-Agirretxe, I.; Vecino, E.; Astigarraga, E.; Acera, A.; Barreda-Gómez, G. Elevation of Tear MMP-9 Concentration as a Biomarker of Inflammation in Ocular Pathology by Antibody Microarray Immunodetection Assays. Int. J. Mol. Sci. 2022, 23, 5639.
- Lennikov, A.; Yang, M.; Chang, K.; Pan, L.; Saddala, M.S.; Lee, C.; Ashok, A.; Cho, K.S.; Utheim, T.P.; Chen, D.F. Direct Modulation of Microglial Function by Electrical Field. Front. Cell Dev. Biol. 2022, 10, 980775.
- Schuhmann, M.K.; Papp, L.; Stoll, G.; Blum, R.; Volkmann, J.; Fluri, F. Mesencephalic Electrical Stimulation Reduces Neuroinflammation after Photothrombotic Stroke in Rats by Targeting the Cholinergic Anti-Inflammatory Pathway. Int. J. Mol. Sci. 2021, 22, 1254.
- Lee, C.Y.P.; Chooi, W.H.; Ng, S.Y.; Chew, S.Y. Modulating Neuroinflammation through Molecular, Cellular and Biomaterial-Based Approaches to Treat Spinal Cord Injury. Bioeng. Transl. Med. 2022, 8, e10389.
- Song, G.; Zhao, M.; Chen, H.; Lenahan, C.; Zhou, X.; Ou, Y.; He, Y. The Role of Nanomaterials in Stroke Treatment: Targeting Oxidative Stress. Oxid. Med. Cell. Longev. 2021, 2021, 8857486.
- Kim, T.H.; Kang, M.S.; Mandakhbayar, N.; El-Fiqi, A.; Kim, H.W. Anti-Inflammatory Actions of Folate-Functionalized Bioactive Ion-Releasing Nanoparticles Imply Drug-Free Nanotherapy of Inflamed Tissues. Biomaterials 2019, 207, 23–38.
- Shcharbina, N.; Shcharbin, D.; Bryszewska, M. Nanomaterials in Stroke Treatment: Perspectives. Stroke 2013, 44, 2351–2355.
- Pottorf, T.S.; Rotterman, T.M.; McCallum, W.M.; Haley-Johnson, Z.A.; Alvarez, F.J. The Role of Microglia in Neuroinflammation of the Spinal Cord after Peripheral Nerve Injury. Cells 2022, 11, 2083.
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol. Psychiatry 2009, 65, 732–741.
- Kölliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An Integrating Overview of Reactive-Neuroimmune Cell Interactions in Health and Disease. Mediat. Inflamm. 2021, 2021, 9999146.
- Shastri, A.; Bonifati, D.M.; Kishore, U. Innate Immunity and Neuroinflammation. Mediators Inflamm. 2013, 2013, 342931.
- Wang, D.S.; Zurek, A.A.; Lecker, I.; Yu, J.; Abramian, A.M.; Avramescu, S.; Davies, P.A.; Moss, S.J.; Lu, W.Y.; Orser, B.A. Memory Deficits Induced by Inflammation Are Regulated by A5-Subunit-Containing GABAA Receptors. Cell Rep. 2012, 2, 488–496.
- Leboyer, M.; Soreca, I.; Scott, J.; Frye, M.; Henry, C.; Tamouza, R.; Kupfer, D.J. Can Bipolar Disorder Be Viewed as a Multi-System Inflammatory Disease? J. Affect. Disord. 2012, 141, 1–10.
- Muller, N.; Myint, A.-M.J.; Schwarz, M. Kynurenine Pathway in Schizophrenia: Pathophysiological and Therapeutic Aspects. Curr. Pharm. Des. 2011, 17, 130–136.
- McNally, L.; Bhagwagar, Z.; Hannestad, J. Inflammation, Glutamate, and Glia in Depression: A Literature Review. CNS Spectr. 2008, 13, 501–510.
- O’Connor, J.C.; André, C.; Wang, Y.; Lawson, M.A.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Interferon-Gamma and Tumor Necrosis Factor-Alpha Mediate the Upregulation of Indoleamine 2,3-Dioxygenase and the Induction of Depressive-like Behavior in Mice in Response to Bacillus Calmette-Guerin. J. Neurosci. 2009, 29, 4200–4209.
- Pedersen, C.C.; Ushakova, A.; Skogseth, R.E.; Alves, G.; Tysnes, O.B.; Aarsland, D.; Lange, J.; Maple-Grødem, J. Inflammatory Biomarkers in Newly Diagnosed Patients With Parkinson Disease and Related Neurodegenerative Disorders. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, e200132.
- Inicio|NINDS Español. Available online: https://espanol.ninds.nih.gov/es (accessed on 5 September 2023).
- Berru Loayza, K.F. Factores Pronósticos de Morbilidad y Secuelas Del Accidente Cerebrovascular En Adultos Mayores; Universidad Católica de Cuenca: Cuenca, Ecuador, 2021.
- Ruiz-Mejía, A.F.; Pérez-Romero, G.E.; Ángel-Macías, M.A.; Ruiz-Mejía, A.F.; Pérez-Romero, G.E.; Ángel-Macías, M.A. Ataque Cerebrovascular Isquémico: Fisiopatología Desde El Sistema Biomédico y Su Equivalente En La Medicina Tradicional China. Rev. Fac. Med. 2017, 65, 137–144.
- Chaves Sell, F. Accidente Vascular Cerebral: ¿es El Accidente Vascular Cerebral Una Enfermedad Tratable? Rev. Costarric. Cardiol. 2000, 2, 27–33.
- Harrison. Principios de Medicina Interna, 21e|AccessMedicina|McGraw Hill Medical. Available online: https://accessmedicina.mhmedical.com/book.aspx?bookID=3118 (accessed on 5 September 2023).
- Teresa, P.; Ribeiro, C.; Rio, S. Manual de Patologia Bucal; FAPERJ: Rio de Janeiro, Brazil, 2013.
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E.; et al. Temporal and Spatial Dynamics of Cerebral Immune Cell Accumulation in Stroke. Stroke 2009, 40, 1849–1857.
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T Lymphocytes and Interferon-Gamma in Ischemic Stroke. Circulation 2006, 113, 2105–2112.
- Walsh, J.T.; Hendrix, S.; Boato, F.; Smirnov, I.; Zheng, J.; Lukens, J.R.; Gadani, S.; Hechler, D.; Gölz, G.; Rosenberger, K.; et al. MHCII-Independent CD4+ T Cells Protect Injured CNS Neurons via IL-4. J. Clin. Investig. 2015, 125, 699–714.
- Selvaraj, U.M.; Ujas, T.A.; Kong, X.; Kumar, A.; Plautz, E.J.; Zhang, S.; Xing, C.; Sudduth, T.L.; Wilcock, D.M.; Turchan-Cholewo, J.; et al. Delayed Diapedesis of CD8 T Cells Contributes to Long-Term Pathology after Ischemic Stroke in Male Mice. Brain Behav. Immun. 2021, 95, 502–513.
- Polazzi, E.; Monti, B. Microglia and Neuroprotection: From in Vitro Studies to Therapeutic Applications. Prog. Neurobiol. 2010, 92, 293–315.
- Naqvi, I.; Hitomi, E.; Leigh, R. Sustained Opening of the Blood-Brain Barrier with Progressive Accumulation of White Matter Hyperintensities Following Ischemic Stroke. Brain Sci. 2019, 9, 16.
- Bernardo-Castro, S.; Sousa, J.A.; Martins, E.; Donato, H.; Nunes, C.; d’Almeida, O.C.; Castelo-Branco, M.; Abrunhosa, A.; Ferreira, L.; Sargento-Freitas, J. The Evolution of Blood–Brain Barrier Permeability Changes after Stroke and Its Implications on Clinical Outcome: A Systematic Review and Meta-Analysis. Int. J. Stroke 2023, 18, 783–794.
- Arba, F.; Leigh, R.; Inzitari, D.; Warach, S.J.; Luby, M.; Lees, K.R. Blood-Brain Barrier Leakage Increases with Small Vessel Disease in Acute Ischemic Stroke. Neurology 2017, 89, 2143–2150.
- Maynard, F.M., Jr.; Bracken, M.B.; Creasey, G.; Ditunno, J.F., Jr.; Donovan, W.H.; Ducker, T.B.; Garber, S.L.; Marino, R.J.; Stover, S.L.; Tator, C.H.; et al. International Standards for Neurological and Functional Classification of Spinal Cord Injury. Spinal Cord 1997, 35, 266–274.
- Hakimi, K.N.; Massagli, T.L. Anterior Spinal Artery Syndrome in Two Children with Genetic Thrombotic Disorders. J. Spinal Cord. Med. 2005, 28, 69–73.
- Roth, E.J.; Park, T.; Pang, T.; Yarkony, G.M.; Lee, M.Y. Traumatic Cervical Brown-Sequard and Brown-Sequard-plus Syndromes: The Spectrum of Presentations and Outcomes. Paraplegia 1991, 29, 582–589.
- McKinley, W.; Hills, A.; Sima, A. Posterior Cord Syndrome: Demographics and Rehabilitation Outcomes. J. Spinal Cord. Med. 2021, 44, 241.
- Cauda Equina Syndrome—Symptoms, Causes, Diagnosis and Treatments. Available online: https://www.aans.org/en/Patients/Neurosurgical-Conditions-and-Treatments/Cauda-Equina-Syndrome (accessed on 6 September 2023).
- Little, J.W.; Ditunno, J.F.; Stiens, S.A.; Harris, R.M. Incomplete Spinal Cord Injury: Neuronal Mechanisms of Motor Recovery and Hyperreflexia. Arch. Phys. Med. Rehabil. 1999, 80, 587–599.
- Singhal, V.; Aggarwal, R. Chapter 11—Spinal Shock. In Complications in Neuroanesthesia; Academic Press: Cambridge, MA, USA, 2017; pp. 89–94.
- Ditunno, J.F.; Little, J.W.; Tessler, A.; Burns, A.S. Spinal Shock Revisited: A Four-Phase Model. Spinal Cord. 2004, 42, 383–395.
- Smith, P.M.; Jeffery, N.D. Spinal Shock—Comparative Aspects and Clinical Relevance. J. Vet. Intern. Med. 2005, 19, 788–793.
- Shik, M.L.; Orlovsky, G.N. Neurophysiology of Locomotor Automatism. Physiol. Rev. 1976, 56, 465–501.
- Min, K.J.; Jeong, H.K.; Kim, B.; Hwang, D.H.; Shin, H.Y.; Nguyen, A.T.; Kim, J.H.; Jou, I.; Kim, B.G.; Joe, E. hye Spatial and Temporal Correlation in Progressive Degeneration of Neurons and Astrocytes in Contusion-Induced Spinal Cord Injury. J. Neuroinflamm. 2012, 9, 100.
- Ji, K.A.; Yang, M.S.; Jeong, H.K.; Min, K.J.; Kang, S.H.; Jou, I.; Joe, E.H. Resident Microglia Die and Infiltrated Neutrophils and Monocytes Become Major Inflammatory Cells in Lipopolysaccharide-Injected Brain. Glia 2007, 55, 1577–1588.
- Pineau, I.; Sun, L.; Bastien, D.; Lacroix, S. Astrocytes Initiate Inflammation in the Injured Mouse Spinal Cord by Promoting the Entry of Neutrophils and Inflammatory Monocytes in an IL-1 Receptor/MyD88-Dependent Fashion. Brain Behav. Immun. 2010, 24, 540–553.
- Stirling, D.P.; Yong, V.W. Dynamics of the Inflammatory Response after Murine Spinal Cord Injury Revealed by Flow Cytometry. J. Neurosci. Res. 2008, 86, 1944–1958.
- Fleming, J.C.; Norenberg, M.D.; Ramsay, D.A.; Dekaban, G.A.; Marcillo, A.E.; Saenz, A.D.; Pasquale-Styles, M.; Dietrich, W.D.; Weaver, L.C. The Cellular Inflammatory Response in Human Spinal Cords after Injury. Brain 2006, 129, 3249–3269.
- Hawkins, B.T.; Davis, T.P. The Blood-Brain Barrier/Neurovascular Unit in Health and Disease. Pharmacol. Rev. 2005, 57, 173–185.
- TIMPL, R. Structure and Biological Activity of Basement Membrane Proteins. Eur. J. Biochem. 1989, 180, 487–502.
- Scholz, M.; Cinatl, J.; Schädel-Höpfner, M.; Windolf, J. Neutrophils and the Blood-Brain Barrier Dysfunction after Trauma. Med. Res. Rev. 2007, 27, 401–416.
- Lee, S.M.; Rosen, S.; Weinstein, P.; Van Rooijen, N.; Noble-Haeusslein, L.J. Prevention of Both Neutrophil and Monocyte Recruitment Promotes Recovery after Spinal Cord Injury. J. Neurotrauma 2011, 28, 1893–1907.
- Hynes, R.O. Integrins: Versatility, Modulation, and Signaling in Cell Adhesion. Cell 1992, 69, 11–25.
- Yang, L.; Jin, P.; Wang, X.; Zhou, Q.; Lin, X.; Xi, S. Fluoride Activates Microglia, Secretes Inflammatory Factors and Influences Synaptic Neuron Plasticity in the Hippocampus of Rats. Neurotoxicology 2018, 69, 108–120.
- Nishimura, Y.; Onoe, H.; Morichika, Y.; Perfiliev, S.; Tsukada, H.; Isa, T. Time-Dependent Central Compensatory Mechanisms of Finger Dexterity after Spinal Cord Injury. Science 2007, 318, 1150–1155.
- Beck, H.; Yaari, Y. Plasticity of Intrinsic Neuronal Properties in CNS Disorders. Nat. Rev. Neurosci. 2008, 9, 357–369.
- Barbizan, R.; Oliveira, A.L.R. Impact of Acute Inflammation on Spinal Motoneuron Synaptic Plasticity Following Ventral Root Avulsion. J. Neuroinflamm. 2010, 7, 29.
- Chen, T.; Yu, Y.; Tang, L.J.; Kong, L.; Zhang, C.H.; Chu, H.Y.; Yin, L.W.; Ma, H.Y. Neural Stem Cells Over-Expressing Brain-Derived Neurotrophic Factor Promote Neuronal Survival and Cytoskeletal Protein Expression in Traumatic Brain Injury Sites. Neural Regen. Res. 2017, 12, 433.
- Chandhok, G.; Lazarou, M.; Neumann, B. Structure, Function, and Regulation of Mitofusin-2 in Health and Disease. Biol. Rev. Camb. Philos. Soc. 2017, 93, 933–949.
- Duann, P.; Lin, P.H. Mitochondria Damage and Kidney Disease. Adv. Exp. Med. Biol. 2017, 982, 529–551.
- Krauss, S. Mitochondria: Structure and Role in Respiration. In eLS (Encyclopedia of Life Sciences); John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2001.
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and Endoplasmic Reticulum Calcium Homeostasis and Cell Death. Cell Calcium 2017, 69, 62–72.
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as Playmakers of Apoptosis, Autophagy and Senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153.
- Giménez-Palomo, A.; Dodd, S.; Anmella, G.; Carvalho, A.F.; Scaini, G.; Quevedo, J.; Pacchiarotti, I.; Vieta, E.; Berk, M. The Role of Mitochondria in Mood Disorders: From Physiology to Pathophysiology and to Treatment. Front. Psychiatry 2021, 12, 546801.
- Van Der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871.
- Yang, B.; Chen, Y.; Shi, J. Reactive Oxygen Species (ROS)-Based Nanomedicine. Chem. Rev. 2019, 119, 4881–4985.
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804.
- Buttgereit, F.; Burmester, G.R.; Brand, M.D. Bioenergetics of Immune Functions: Fundamental and Therapeutic Aspects. Immunol. Today 2000, 21, 194–199.
- Kwon, H.S.; Koh, S.H. Neuroinflammation in Neurodegenerative Disorders: The Roles of Microglia and Astrocytes. Transl. Neurodegener. 2020, 9, 42.
- Felger, J.C.; Lotrich, F.E. Inflammatory Cytokines in Depression: Neurobiological Mechanisms and Therapeutic Implications. Neuroscience 2013, 246, 199–229.
- Blanco, L.P.; Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832.
- Casaril, A.M.; Dantzer, R.; Bas-Orth, C. Neuronal Mitochondrial Dysfunction and Bioenergetic Failure in Inflammation-Associated Depression. Front. Neurosci. 2021, 15, 725547.
- Wang, X.J.; Kong, K.M.; Qi, W.L.; Ye, W.L.; Song, P.S. Interleukin-1 Beta Induction of Neuron Apoptosis Depends on P38 Mitogen-Activated Protein Kinase Activity after Spinal Cord Injury. Acta Pharmacol. Sin. 2005, 26, 934–942.
- Hellenbrand, D.J.; Quinn, C.M.; Piper, Z.J.; Morehouse, C.N.; Fixel, J.A.; Hanna, A.S. Inflammation after Spinal Cord Injury: A Review of the Critical Timeline of Signaling Cues and Cellular Infiltration. J. Neuroinflamm. 2021, 18, 284.
- Hamza, T.; Barnett, J.B.; Li, B. Interleukin 12 a Key Immunoregulatory Cytokine in Infection Applications. Int. J. Mol. Sci. 2010, 11, 789–806.
- Hill, F.; Kim, C.F.; Gorrie, C.A.; Moalem-Taylor, G. Interleukin-17 Deficiency Improves Locomotor Recovery and Tissue Sparing after Spinal Cord Contusion Injury in Mice. Neurosci. Lett. 2011, 487, 363–367.
- Onishi, R.M.; Gaffen, S.L. Interleukin-17 and Its Target Genes: Mechanisms of Interleukin-17 Function in Disease. Immunology 2010, 129, 311–321.
- Ousman, S.S.; David, S. MIP-1alpha, MCP-1, GM-CSF, and TNF-α Control the Immune Cell Response That Mediates Rapid Phagocytosis of Myelin from the Adult Mouse Spinal Cord. J. Neurosci. 2001, 21, 4649–4656.
- Shen, H.; Xu, B.; Yang, C.; Xue, W.; You, Z.; Wu, X.; Ma, D.; Shao, D.; Leong, K.; Dai, J. A DAMP-Scavenging, IL-10-Releasing Hydrogel Promotes Neural Regeneration and Motor Function Recovery after Spinal Cord Injury. Biomaterials 2022, 280, 121279.
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 338745.
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The Adenosine-Dependent Angiogenic Switch of Macrophages to an M2-like Phenotype Is Independent of Interleukin-4 Receptor Alpha (IL-4Rα) Signaling. Inflammation 2013, 36, 921–931.
- Lin, S.; Xu, C.; Lin, J.; Hu, H.; Zhang, C.; Mei, X. Regulation of Inflammatory Cytokines for Spinal Cord Injury Recovery. Histol. Histopathol. 2021, 36, 137–142.
- Yang, L.; Blumbergs, P.C.; Jones, N.R.; Manavis, J.; Sarvestani, G.T.; Ghabriel, M.N. Early Expression and Cellular Localization of Proinflammatory Cytokines Interleukin-1beta, Interleukin-6, and Tumor Necrosis Factor-Alpha in Human Traumatic Spinal Cord Injury. Spine 2004, 29, 966–971.
- Garcia, E.; Aguilar-Cevallos, J.; Silva-Garcia, R.; Ibarra, A. Cytokine and Growth Factor Activation In Vivo and In Vitro after Spinal Cord Injury. Mediat. Inflamm. 2016, 2016, 9476020.
- Lee, H.J.; Kim, C.; Lee, S.J. Alpha-Synuclein Stimulation of Astrocytes: Potential Role for Neuroinflammation and Neuroprotection. Oxid. Med. Cell. Longev. 2010, 3, 283–287.
- Ransohoff, R.M. The Chemokine System in Neuroinflammation: An Update. J. Infect. Dis. 2002, 186, S152–S156.
- Kiguchi, N.; Kobayashi, Y.; Kishioka, S. Chemokines and Cytokines in Neuroinflammation Leading to Neuropathic Pain. Curr. Opin. Pharmacol. 2012, 12, 55–61.
- Ubogu, E.E.; Callahan, M.K.; Tucky, B.H.; Ransohoff, R.M. Determinants of CCL5-Driven Mononuclear Cell Migration across the Blood–Brain Barrier. Implications for Therapeutically Modulating Neuroinflammation. J. Neuroimmunol. 2006, 179, 132–144.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
791
Revisions:
2 times
(View History)
Update Date:
02 Nov 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No