 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zhili Dong | -- | 1939 | 2023-11-01 14:35:56 | | | |
| 2 | Wendy Huang | Meta information modification | 1939 | 2023-11-02 02:28:41 | | |
Video Upload Options
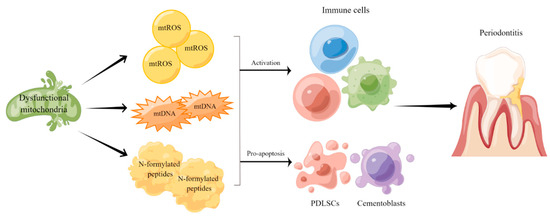
Oral inflammatory diseases (OIDs) include many common diseases such as periodontitis and pulpitis. The causes of OIDs consist microorganism, trauma, occlusal factors, autoimmune dis-eases and radiation therapy. When treated unproperly, such diseases not only affect oral health but also pose threat to people’s overall health condition. Therefore, identifying OIDs at an early stage and exploring new therapeutic strategies are important tasks for oral-related research. Mitochondria are crucial organelles for many cellular activities and disruptions of mitochondrial function not only affect cellular metabolism but also indirectly influence people’s health and life span. Increasing evidence suggests that mitochondrial dysfunction plays a critical role in the development and progression of OIDs and its associated systemic diseases.
1. Introduction
2. The Role of Mitochondrial Dysfunction in the Etiopathogenesis of the Chronic Periodontitis
3. Mitochondrial Dysfunction-Targeted Therapies
References
- Li, X.; Liu, X.C.; Ding, X.; Liu, X.M.; Cao, N.B.; Deng, Y.; Hou, Y.B.; Yu, W.X. Resveratrol protects renal damages induced by periodontitis via preventing mitochondrial dysfunction in rats. Oral. Dis. 2022, 29, 1812–1825.
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218.
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153.
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754.
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259.
- Sangwung, P.; Petersen, K.F.; Shulman, G.I.; Knowles, J.W. Mitochondrial Dysfunction, Insulin Resistance, and Potential Genetic Implications. Endocrinology 2020, 161, bqaa017.
- Moro, L. Mitochondrial Dysfunction in Aging and Cancer. J. Clin. Med. 2019, 8, 1983.
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863.
- Gong, W.; Wang, F.; He, Y.; Zeng, X.; Zhang, D.; Chen, Q. Mesenchymal Stem Cell Therapy for Oral Inflammatory Diseases: Research Progress and Future Perspectives. Curr. Stem Cell Res. Ther. 2021, 16, 165–174.
- Bitencourt, F.V.; Nascimento, G.G.; Costa, S.A.; Andersen, A.; Sandbæk, A.; Leite, F.R.M. Co-occurrence of Periodontitis and Diabetes-Related Complications. J. Dent. Res. 2023, 102, 1088–1097.
- Vujovic, S.; Desnica, J.; Stevanovic, M.; Mijailovic, S.; Vojinovic, R.; Selakovic, D.; Jovicic, N.; Rosic, G.; Milovanovic, D. Oral Health and Oral Health-Related Quality of Life in Patients with Primary Sjögren’s Syndrome. Medicina 2023, 59, 473.
- Sødal, A.T.T.; Skudutyte-Rysstad, R.; Diep, M.T.; Koldsland, O.C.; Hove, L.H. Periodontitis in a 65-year-old population: Risk indicators and impact on oral health-related quality of life. BMC Oral Health 2022, 22, 640.
- Taha, N.A.; Abuzaid, A.M.; Khader, Y.S. A Randomized Controlled Clinical Trial of Pulpotomy versus Root Canal Therapy in Mature Teeth with Irreversible Pulpitis: Outcome, Quality of Life, and Patients’ Satisfaction. J. Endod. 2023, 49, 624–631.e622.
- Jiang, W.; Wang, Y.; Cao, Z.; Chen, Y.; Si, C.; Sun, X.; Huang, S. The role of mitochondrial dysfunction in periodontitis: From mechanisms to therapeutic strategy. J. Periodontal Res. 2023, 58, 853–863.
- Verma, A.; Azhar, G.; Zhang, X.; Patyal, P.; Kc, G.; Sharma, S.; Che, Y.; Wei, J.Y.P. gingivalis-LPS Induces Mitochondrial Dysfunction Mediated by Neuroinflammation through Oxidative Stress. Int. J. Mol. Sci. 2023, 24, 950.
- Zhou, L.; Zhang, Y.F.; Yang, F.H.; Mao, H.Q.; Chen, Z.; Zhang, L. Mitochondrial DNA leakage induces odontoblast inflammation via the cGAS-STING pathway. Cell Commun. Signal 2021, 19, 58.
- Demmer, R.T.; Papapanou, P.N. Epidemiologic patterns of chronic and aggressive periodontitis. Periodontol. 2000 2010, 53, 28–44.
- Genco, R.J.; Sanz, M. Clinical and public health implications of periodontal and systemic diseases: An overview. Periodontol. 2000 2020, 83, 7–13.
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Periodontol. 2018, 89 (Suppl. S1), S173–S182.
- Laine, M.L.; Crielaard, W.; Loos, B.G. Genetic susceptibility to periodontitis. Periodontol. 2000 2012, 58, 37–68.
- Tóthová, L.; Celec, P. Oxidative Stress and Antioxidants in the Diagnosis and Therapy of Periodontitis. Front. Physiol. 2017, 8, 1055.
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal diseases. Nat. Rev. Dis. Primers 2017, 3, 17038.
- Genco, R.J.; Borgnakke, W.S. Risk factors for periodontal disease. Periodontol. 2000 2013, 62, 59–94.
- Graziani, F.; Karapetsa, D.; Alonso, B.; Herrera, D. Nonsurgical and surgical treatment of periodontitis: How many options for one disease? Periodontol. 2000 2017, 75, 152–188.
- Kwon, T.; Lamster, I.B.; Levin, L. Current Concepts in the Management of Periodontitis. Int. Dent. J. 2021, 71, 462–476.
- Prietto, N.R.; Martins, T.M.; Santinoni, C.D.S.; Pola, N.M.; Ervolino, E.; Bielemann, A.M.; Leite, F.R.M. Treatment of experimental periodontitis with chlorhexidine as adjuvant to scaling and root planing. Arch. Oral. Biol. 2020, 110, 104600.
- Xie, M.; Tang, Q.; Nie, J.; Zhang, C.; Zhou, X.; Yu, S.; Sun, J.; Cheng, X.; Dong, N.; Hu, Y.; et al. BMAL1-Downregulation Aggravates Porphyromonas Gingivalis-Induced Atherosclerosis by Encouraging Oxidative Stress. Circ. Res. 2020, 126, e15–e29.
- Bullon, P.; Newman, H.N.; Battino, M. Obesity, diabetes mellitus, atherosclerosis and chronic periodontitis: A shared pathology via oxidative stress and mitochondrial dysfunction? Periodontol. 2000 2014, 64, 139–153.
- Li, L.; Zhang, Y.L.; Liu, X.Y.; Meng, X.; Zhao, R.Q.; Ou, L.L.; Li, B.Z.; Xing, T. Periodontitis Exacerbates and Promotes the Progression of Chronic Kidney Disease through Oral Flora, Cytokines, and Oxidative Stress. Front. Microbiol. 2021, 12, 656372.
- Govindaraj, P.; Khan, N.A.; Gopalakrishna, P.; Chandra, R.V.; Vanniarajan, A.; Reddy, A.A.; Singh, S.; Kumaresan, R.; Srinivas, G.; Singh, L.; et al. Mitochondrial dysfunction and genetic heterogeneity in chronic periodontitis. Mitochondrion 2011, 11, 504–512.
- Tomokiyo, A.; Wada, N.; Maeda, H. Periodontal Ligament Stem Cells: Regenerative Potency in Periodontium. Stem Cells Dev. 2019, 28, 974–985.
- Zhang, Z.; Deng, M.; Hao, M.; Tang, J. Periodontal ligament stem cells in the periodontitis niche: Inseparable interactions and mechanisms. J. Leukoc. Biol. 2021, 110, 565–576.
- Zheng, Y.; Dong, C.; Yang, J.; Jin, Y.; Zheng, W.; Zhou, Q.; Liang, Y.; Bao, L.; Feng, G.; Ji, J.; et al. Exosomal microRNA-155-5p from PDLSCs regulated Th17/Treg balance by targeting sirtuin-1 in chronic periodontitis. J. Cell Physiol. 2019, 234, 20662–20674.
- Li, J.; Wang, Z.; Huang, X.; Wang, Z.; Chen, Z.; Wang, R.; Chen, Z.; Liu, W.; Wu, B.; Fang, F.; et al. Dynamic proteomic profiling of human periodontal ligament stem cells during osteogenic differentiation. Stem Cell Res. Ther. 2021, 12, 98.
- Chen, Y.; Ji, Y.; Jin, X.; Sun, X.; Zhang, X.; Chen, Y.; Shi, L.; Cheng, H.; Mao, Y.; Li, X.; et al. Mitochondrial abnormalities are involved in periodontal ligament fibroblast apoptosis induced by oxidative stress. Biochem. Biophys. Res. Commun. 2019, 509, 483–490.
- Liu, J.; Zeng, J.; Wang, X.; Zheng, M.; Luan, Q. P53 mediates lipopolysaccharide-induced inflammation in human gingival fibroblasts. J. Periodontol. 2018, 89, 1142–1151.
- Liu, J.; Wang, X.; Xue, F.; Zheng, M.; Luan, Q. Abnormal mitochondrial structure and function are retained in gingival tissues and human gingival fibroblasts from patients with chronic periodontitis. J. Periodontal Res. 2022, 57, 94–103.
- Liu, J.; Wang, X.; Zheng, M.; Luan, Q. Oxidative stress in human gingival fibroblasts from periodontitis versus healthy counterparts. Oral. Dis. 2021, 29, 1214–1225.
- França, L.F.C.; Vasconcelos, A.; da Silva, F.R.P.; Alves, E.H.P.; Carvalho, J.S.; Lenardo, D.D.; de Souza, L.K.M.; Barbosa, A.L.R.; Medeiros, J.R.; de Oliveira, J.S.; et al. Periodontitis changes renal structures by oxidative stress and lipid peroxidation. J. Clin. Periodontol. 2017, 44, 568–576.
- Kose, O.; Kurt Bayrakdar, S.; Unver, B.; Altin, A.; Akyildiz, K.; Mercantepe, T.; Bostan, S.A.; Arabaci, T.; Turker Sener, L.; Emre Kose, T.; et al. Melatonin improves periodontitis-induced kidney damage by decreasing inflammatory stress and apoptosis in rats. J. Periodontol. 2021, 92, 22–34.
- Sun, X.; Mao, Y.; Dai, P.; Li, X.; Gu, W.; Wang, H.; Wu, G.; Ma, J.; Huang, S. Mitochondrial dysfunction is involved in the aggravation of periodontitis by diabetes. J. Clin. Periodontol. 2017, 44, 463–471.
- Zhang, B.; Yang, Y.; Yi, J.; Zhao, Z.; Ye, R. Hyperglycemia modulates M1/M2 macrophage polarization via reactive oxygen species overproduction in ligature-induced periodontitis. J. Periodontal Res. 2021, 56, 991–1005.
- Zeng, X.T.; Leng, W.D.; Lam, Y.Y.; Yan, B.P.; Wei, X.M.; Weng, H.; Kwong, J.S. Periodontal disease and carotid atherosclerosis: A meta-analysis of 17,330 participants. Int. J. Cardiol. 2016, 203, 1044–1051.
- Priyamvara, A.; Dey, A.K.; Bandyopadhyay, D.; Katikineni, V.; Zaghlol, R.; Basyal, B.; Barssoum, K.; Amarin, R.; Bhatt, D.L.; Lavie, C.J. Periodontal Inflammation and the Risk of Cardiovascular Disease. Curr. Atheroscler. Rep. 2020, 22, 28.
- Rho, J.H.; Kim, H.J.; Joo, J.Y.; Lee, J.Y.; Lee, J.H.; Park, H.R. Periodontal Pathogens Promote Foam Cell Formation by Blocking Lipid Efflux. J. Dent. Res. 2021, 100, 1367–1377.
- Xu, T.; Dong, Q.; Luo, Y.; Liu, Y.; Gao, L.; Pan, Y.; Zhang, D. Porphyromonas gingivalis infection promotes mitochondrial dysfunction through Drp1-dependent mitochondrial fission in endothelial cells. Int. J. Oral. Sci. 2021, 13, 28.
- Liu, Q.; Guo, S.; Huang, Y.; Wei, X.; Liu, L.; Huo, F.; Huang, P.; Wu, Y.; Tian, W. Inhibition of TRPA1 Ameliorates Periodontitis by Reducing Periodontal Ligament Cell Oxidative Stress and Apoptosis via PERK/eIF2α/ATF-4/CHOP Signal Pathway. Oxid. Med. Cell Longev. 2022, 2022, 4107915.
- Gölz, L.; Memmert, S.; Rath-Deschner, B.; Jäger, A.; Appel, T.; Baumgarten, G.; Götz, W.; Frede, S. Hypoxia and P. gingivalis synergistically induce HIF-1 and NF-κB activation in PDL cells and periodontal diseases. Mediat. Inflamm. 2015, 2015, 438085.
- Zhao, J.; Faure, L.; Adameyko, I.; Sharpe, P.T. Stem cell contributions to cementoblast differentiation in healthy periodontal ligament and periodontitis. Stem Cells 2021, 39, 92–102.
- Arzate, H.; Zeichner-David, M.; Mercado-Celis, G. Cementum proteins: Role in cementogenesis, biomineralization, periodontium formation and regeneration. Periodontol. 2000 2015, 67, 211–233.
- Wang, H.; Wang, X.; Ma, L.; Huang, X.; Peng, Y.; Huang, H.; Gao, X.; Chen, Y.; Cao, Z. PGC-1 alpha regulates mitochondrial biogenesis to ameliorate hypoxia-inhibited cementoblast mineralization. Ann. N.Y. Acad. Sci. 2022, 1516, 300–311.
- Zhao, B.; Zhang, W.; Xiong, Y.; Zhang, Y.; Zhang, D.; Xu, X. Effects of rutin on the oxidative stress, proliferation and osteogenic differentiation of periodontal ligament stem cells in LPS-induced inflammatory environment and the underlying mechanism. J. Mol. Histol. 2020, 51, 161–171.
- Iova, G.M.; Calniceanu, H.; Popa, A.; Szuhanek, C.A.; Marcu, O.; Ciavoi, G.; Scrobota, I. The Antioxidant Effect of Curcumin and Rutin on Oxidative Stress Biomarkers in Experimentally Induced Periodontitis in Hyperglycemic Wistar Rats. Molecules 2021, 26, 1332.
- Cai, W.J.; Chen, Y.; Shi, L.X.; Cheng, H.R.; Banda, I.; Ji, Y.H.; Wang, Y.T.; Li, X.M.; Mao, Y.X.; Zhang, D.F.; et al. AKT-GSK3β Signaling Pathway Regulates Mitochondrial Dysfunction-Associated OPA1 Cleavage Contributing to Osteoblast Apoptosis: Preventative Effects of Hydroxytyrosol. Oxid. Med. Cell Longev. 2019, 2019, 4101738.
- Zhang, X.; Jiang, Y.; Mao, J.; Ren, X.; Ji, Y.; Mao, Y.; Chen, Y.; Sun, X.; Pan, Y.; Ma, J.; et al. Hydroxytyrosol prevents periodontitis-induced bone loss by regulating mitochondrial function and mitogen-activated protein kinase signaling of bone cells. Free Radic. Biol. Med. 2021, 176, 298–311.
- Jiang, C.; Yang, W.; Wang, C.; Qin, W.; Ming, J.; Zhang, M.; Qian, H.; Jiao, T. Methylene Blue-Mediated Photodynamic Therapy Induces Macrophage Apoptosis via ROS and Reduces Bone Resorption in Periodontitis. Oxid. Med. Cell Longev. 2019, 2019, 1529520.
- Chambrone, L.; Wang, H.L.; Romanos, G.E. Antimicrobial photodynamic therapy for the treatment of periodontitis and peri-implantitis: An American Academy of Periodontology best evidence review. J. Periodontol. 2018, 89, 783–803.
- Sui, L.; Wang, J.; Xiao, Z.; Yang, Y.; Yang, Z.; Ai, K. ROS-Scavenging Nanomaterials to Treat Periodontitis. Front. Chem. 2020, 8, 595530.
- Mok, Z.H.; Proctor, G.; Thanou, M. Emerging nanomaterials for dental treatments. Emerg. Top. Life Sci. 2020, 4, 613–625.
- Ren, S.; Zhou, Y.; Fan, R.; Peng, W.; Xu, X.; Li, L.; Xu, Y. Constructing biocompatible MSN@Ce@PEG nanoplatform for enhancing regenerative capability of stem cell via ROS-scavenging in periodontitis. Chem. Eng. J. 2021, 423, 130207.
- Li, X.; Zhao, Y.; Peng, H.; Gu, D.; Liu, C.; Ren, S.; Miao, L. Robust intervention for oxidative stress-induced injury in periodontitis via controllably released nanoparticles that regulate the ROS-PINK1-Parkin pathway. Front. Bioeng. Biotechnol. 2022, 10, 1081977.
- Qiu, X.; Yu, Y.; Liu, H.; Li, X.; Sun, W.; Wu, W.; Liu, C.; Miao, L. Remodeling the periodontitis microenvironment for osteogenesis by using a reactive oxygen species-cleavable nanoplatform. Acta Biomater. 2021, 135, 593–605.
- Zhai, Q.; Chen, X.; Fei, D.; Guo, X.; He, X.; Zhao, W.; Shi, S.; Gooding, J.J.; Jin, F.; Jin, Y.; et al. Nanorepairers Rescue Inflammation-Induced Mitochondrial Dysfunction in Mesenchymal Stem Cells. Adv. Sci. 2022, 9, e2103839.
- Nessa, N.; Kobara, M.; Toba, H.; Adachi, T.; Yamamoto, T.; Kanamura, N.; Pezzotti, G.; Nakata, T. Febuxostat Attenuates the Progression of Periodontitis in Rats. Pharmacology 2021, 106, 294–304.

