A mitochondrion is a double membrane-enclosed organelle found in most eukaryotic cells, with a size ranging from 0.5 to 10 μm in diameter and the number of mitochondria per cell varying according to energy demand. Mitochondria are often referred to as the cellular power plants or energy center because they generate most of the adenosine-5′-triphosphate (ATP) via oxidative phosphorylation (OXPHOS) as the major source of chemical energy for physiological processes. Since mitochondria generate most reactive oxygen species (ROS) via OXPHOS and possess effective antioxidant systems, they play a central role in regulating oxidative stress and cellular redox homeostasis [
1,
2]. In addition to generating cellular energy, mitochondria are involved in many other processes, such as lipid metabolism, cellular differentiation, immune regulation, apoptosis, autophagy, cell growth and protein synthesis [
3,
4,
5]. Given that mitochondria play pleiotropic roles in cellular physiology, mitochondrial dysfunction can directly regulate cell and tissue homeostasis and participate in the pathological process of many systemic diseases such as diabetes mellitus, cancer and autoimmune diseases [
6,
7,
8].
2. The Role of Mitochondrial Dysfunction in the Etiopathogenesis of the Chronic Periodontitis
As one of the most common human diseases, periodontitis is a chronic inflammatory disease that affects about 45% of adults, rising to over 60% in people aged over 65, which creates a significant healthcare, social and economic burden when left untreated or not treated appropriately [
48,
49]. Common features of periodontitis include gingival inflammation, clinical attachment loss and alveolar bone loss [
50]. Although the main causative factor is microorganisms which colonize the subgingival dental plaque—inducing an exaggerated inflammatory response—genetic predisposition, smoking, poor oral hygiene and malnutrition are also important factors in the pathogenesis and progression of periodontitis [
51,
52,
53,
54]. Despite recent advances in the understanding of the pathological process, common treatments for periodontitis, including basic treatment, periodontal surgery and adjuvant drug administration, still provide insufficient periodontal tissue repair [
55,
56,
57]. Recently, many studies have suggested that mitochondrial dysfunction could also contribute to the initiation of periodontitis and increase the risk of its related systemic diseases [
52,
58,
59,
60]. Govindaraj et al. conducted a study focusing on mitochondrial dysfunction in the periodontal tissue of chronic periodontitis (CP) patients and found that compared to healthy subjects, mitochondrial membrane potential and oxygen consumption rate of gingival cells from CP patients were reduced by four- and five-fold, respectively, whereas, ROS production was increased by 18%. Moreover, mitochondrial DNA sequencing revealed 14 mutations existed only in periodontal tissues but not in circulation, suggesting that mitochondrial dysfunction and genetic heterogeneity could contribute to the pathogenesis of CP [
61]. These studies highlighted the value of mitochondrial function analysis in the early diagnosis of periodontitis.
Periodontal ligament stem cells (PDLSCs) are a kind of somatic mesenchymal stromal cell (MSC) which show typical mesenchymal stromal cell properties, such as self-renewal, multilineage differentiation and immunoregulation, which are essential for homeostasis in periodontal tissue [
62]. Notably, PDLSCs are impaired under periodontitis and are involved in the progression of inflammation by aggravating immune response and stimulating osteoclast differentiation [
63,
64]. Li et al. adopted a quantitative proteomic technique to investigate the protein expression pattern during human PDLSCs osteogenesis. They found that protein related to OXPHOS may be essential in the osteogenesis process, which highlighted the role of mitochondria in regulating PDLSCs’ differentiation ability [
65]. Chen et al. found that mitochondria abnormalities are present in the oxidative stress-induced periodontal ligament fibroblast apoptosis, judging by the increased mtROS amounts, upregulated mitochondrial membrane potential and ATP production [
66]. Liu et al. showed that LPS-induced inflammatory responses in human gingival fibroblasts (HGFs) were partially dependent on the interaction between P53 and ROS. Upon activation, P53 and ROS formed a feedback loop and led to disrupted redox imbalance and mitochondrial dysfunction in periodontitis, triggering increased secretion of IL-1
β, IL-6 and tumor necrosis factor (TNF)-
α [
67]. Liu et al. collected a gingiva tissue sample from CP patients and observed greater mitochondrial structure destruction, reduced mtDNA copy and lower mitochondrial protein PDK2 levels, compared with those samples from healthy individuals [
68]. Similarly, HGFs from periodontitis patients exhibited increased levels of mitochondrial p53, enhanced mtROS production and secretion of pro-inflammatory cytokines, as compared to HGFs from healthy donors [
69].
In an experimental periodontitis rat model, Franca et al. observed morphometric changes in renal tissues and disruption of the brush border in renal tubules, accompanied by an increase in oxidative stress and lipid peroxidation in kidneys [
70]. In another study, melatonin treatment in periodontitis rats was found to be effective in restoring redox balance in gingival tissue and reducing oxidative stress levels in circulation, which could alleviate kidney injury [
71]. Sun et al. observed that diabetic rats with periodontitis presented more severe mitochondrial dysfunction than non-diabetic rats with periodontitis, reflected by the decreased ATP production, reduced gene expression of electron transport chain complex I subunits and weaker mitochondrial biogenesis. They demonstrated a close correlation between these mitochondrial events and periodontal tissue damage, proving that impaired mitochondrial function contributed to the pathogenesis of periodontitis in diabetic rats [
72]. Another study found that diabetic rats displayed enhanced macrophages infiltration and M1 polarization in periodontal lesions, compared with vehicle-treated rats. Under LPS or IL-4 stimulation, RAW264.7 macrophage cells showed elevated ROS levels and increased expression of M1 macrophage markers, which could be reversed by N-acetylcysteine treatment [
73].
Atherosclerosis (AS) is a chronic artery disease characterized by plaque formation and chronic vascular inflammation. Many epidemiological studies have described the correlation between periodontitis and carotid AS [
74,
75]. Porphyromonas gingivalis (
P. gingivalis), a well-known pathogen in periodontitis progression, has been shown to accelerate lipid droplet accumulation in macrophages, partially through the induction of ROS production, leading to disturbed lipid homeostasis and foam cell formation during AS [
76].
P. gingivalis infection can also induce mitochondrial fragmentation, disrupt redox balance and decrease ATP concentration in vascular endothelial cells. Researchers suggested that the phosphorylation and recruitment of Drp1, a key protein involved in mitochondrial fission, might be the key events leading to mitochondrial dysfunction in
P.
gingivalis-infected endothelial cells, providing new insights into how periodontal pathogen-induced mitochondrial dysfunction exacerbates atherosclerotic lesions [
77].
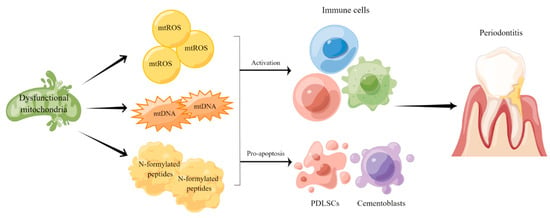
Taken together, the findings mentioned above indicate that mitochondrial dysfunction participates in the pathogenesis and progression of periodontitis by affecting oxidative stress and regulating inflammatory responses (Figure 1). Therefore, developing novel strategies to evaluate mitochondrial function in periodontitis patients may assist the diagnosis and treatment of such disease as well as its complications.
Figure 1. Mitochondrial dysfunction in the pathogenesis and progression of periodontitis. Mitochondrial dysfunction leads to metabolic disruptions and promotes the release of mtROS, mtDNA and N-formylated peptides to regulate immune responses and inflammation in periodontal tissue.
3. Mitochondrial Dysfunction-Targeted Therapies
Liu et al. identified TRPA1, an important transient receptor potential (TRP) cation channel, as an important factor in periodontium destruction in periodontitis. Inhibiting TRPA1 markedly reduced oxidative stress and apoptotic levels in LPS-treated PDLSCs, via the inhibition of endoplasmic reticulum (ER), and mitochondria stress, via downregulating PERK/eIF2α/ATF-4/CHOP pathways [
78]. Periodontitis is closely related to hypoxic microenvironment. Previous studies have demonstrated that oxygen saturation in periodontal microenvironment reduced by 6% [
79]. Cementoblasts possess similar characteristics with osteoblasts and generate cementum in the reconstruction process of periodontal tissue [
80,
81]. Wang et al. showed that overexpression of peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α), a critical regulator of mitochondrial biogenesis, can partially reverse the inhibition of cementoblasts mineralization and mitochondrial biogenesis caused by CoCl
2-induced hypoxia [
82].
Since oxidative stress is recognized as one of the key regulators in periodontitis, the therapeutic efficacy of several antioxidants on periodontitis are examined. Zhao et al. showed that rutin treatment inhibited the release of ROS, increased the secretion of antioxidative factors and promoted PDLSCs proliferation via the PIK3/AKT signaling pathway under an inflammatory environment [
83]. Similar effects were also observed on hyperglycemic periodontitis rats [
84]. Hydroxytyrosol (HT), a natural phenolic compound possessing antioxidative abilities, could inhibit mitochondrial dysfunction by decreasing optic atrophy 1 (OPA1) cleavage and by elevating AKT and GSK3β phosphorylation, which helped prevent oxidative stress-induced osteoblast apoptosis [
85]. HT was also reported to exert a therapeutic effect on the periodontitis mice model via repressing RANKL-induced osteoclast maturation and promoting osteogenic differentiation. Such effect was partly dependent on attenuating mitochondrial dysfunction and inhibiting ERK and JNK pathways [
86]. In addition to antioxidants, photodynamic therapy (PDT) has shown a protective effect on periodontitis by targeting mitochondria as well. Jiang et al. found methylene blue-mediated PDT could induce macrophage apoptosis in vitro and in rats with periodontitis via regulating ROS levels and reducing mitochondrial-dependent apoptosis, suggesting that the potential of PDT in treating periodontitis does not only rely on its antimicrobial capacity [
87,
88].
Recently, nanomaterials showed great potential in the field of periodontitis therapy [
89,
90]. Ren et al. synthesized a nanocomposite with ROS-scavenging activity by combining CeO
2 nanoparticles (CeO
2 NPs) onto the surface of mesoporous silica. Periodontal administration of such nanoparticles efficiently reduced ROS levels and improved the osteogenic differentiative capacity of hPDLSCs with H
2O
2-induced oxidative stress injury [
91]. The same group synthesized controlled drug release nanoparticles by encasing mitoquinone (MitoQ, an autophagy enhancer) into tailor-made ROS-cleavable amphiphilic polymer nanoparticles. Once exposed to ROS under oxidative stress conditions, the ROS-cleavable structure disintegrated, promoting the release of the encapsulated MitoQ. The released MitoQ efficiently induced mitophagy through the PINK1-Parkin pathway and reduced oxidative stress, which contributed to a redox homeostasis and facilitated periodontal tissue regeneration [
92]. Qiu et al. fabricated a ROS-cleavable nanoplatform by encapsulating N-acetylcysteine into tailor-made amphiphilic polymer nanoparticles which could decrease osteoclast activity and inflammation in the periodontitis rat model and improve the restoration of destroyed periodontal tissue [
93]. Zhai et al. showed that obstructed mitophagy and Ca
2+ overload led to dysfunctional mitochondria accumulation in MSCs isolated from periodontitis and osteoarthritis patients. They engineered mitochondria-targeting and intracellular microenvironment-responsive nanoparticles to attract Ca
2+ around mitochondria in MSCs to regulate calcium flux into mitochondria, which successfully restored the mitochondrial function of diseased MSCs and rescued periodontal tissue damage [
94].
In addition to alleviating periodontitis-induced periodontal tissue damage, targeting mitochondrial dysfunction in periodontitis also contributes to ameliorating the complications of periodontitis. Li et al. demonstrated that resveratrol could prevent periodontal tissue destruction, as indicated by the improvement in pocket depth, gingival bleeding and tooth mobility. Meanwhile, resveratrol administration also alleviated periodontitis-induced kidney injury by means of decreasing oxidative stress, regulating mitochondrial membrane potential and increasing mtDNA such as Sirtuin 1 and PGC-1α [
1]. Febuxostat, a potent xanthine oxidase inhibitor, has been shown to attenuate the progression of periodontitis in rats by reducing inflammatory cytokine levels and oxidative stress. It also attenuated periodontitis-induced glucose intolerance and blood pressure elevations, suggesting its therapeutic potential in treating patients with both diabetes and periodontitis [
95].