 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Palma Rocchi | -- | 2187 | 2023-10-30 10:17:36 | | | |
| 2 | Alfred Zheng | Meta information modification | 2187 | 2023-10-31 02:41:18 | | |
Video Upload Options
Prostate cancer (PC) is the second most common cancer in men worldwide. Despite recent advances in diagnosis and treatment, castration-resistant prostate cancer (CRPC) remains a significant medical challenge. Prostate cancer cells can develop mechanisms to resist androgen deprivation therapy, such as AR overexpression, AR mutations, alterations in AR coregulators, increased steroidogenic signaling pathways, outlaw pathways, and bypass pathways. Various treatment options for CRPC exist, including androgen deprivation therapy, chemotherapy, immunotherapy, localized or systemic therapeutic radiation, and PARP inhibitors. However, more research is needed to combat CRPC effectively. Further investigation into the underlying mechanisms of the disease and the development of new therapeutic strategies will be crucial in improving patient outcomes.
1. Introduction
2. Mechanisms Underlying Castration-Resistant Prostate Cancer
2.1. Androgen Receptor Overexpression
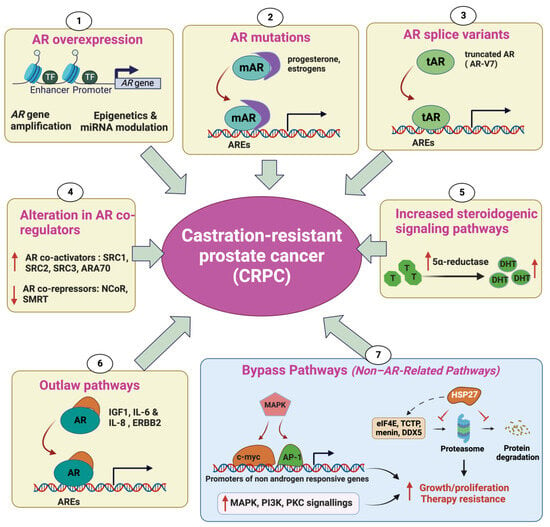
2.2. AR Mutations
2.3. Alteration in AR CoRegulators
2.4. Increased Steroidogenic Signaling Pathways
2.5. Outlaw Pathways
2.6. Non-AR-Related Pathways: Bypass Pathways
2.7. Evolution of t-NEPC from CRPC
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386.
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA. Cancer J. Clin. 2023, 73, 17–48.
- Scher, H.I.; Morris, M.J.; Stadler, W.M.; Higano, C.S.; Halabi, S.; Smith, M.R.; Basch, E.M.; Fizazi, K.; Ryan, C.J.; Antonarakis, E.S.; et al. The Prostate Cancer Working Group 3 (PCWG3) Consensus for Trials in Castration-Resistant Prostate Cancer (CRPC). J. Clin. Oncol. 2015, 33, 5000.
- Castration-Resistant Prostate Cancer: Mechanisms, Targets and Treatment. SpringerLink. Available online: https://link.springer.com/chapter/10.1007/978-3-319-99286-0_7 (accessed on 24 May 2023).
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New Response Evaluation Criteria in Solid Tumours: Revised RECIST Guideline (Version 1.1). Eur. J. Cancer 2009, 45, 228–247.
- Iannantuono, G.M.; Chandran, E.; Floudas, C.S.; Choo-Wosoba, H.; Butera, G.; Roselli, M.; Gulley, J.L.; Karzai, F. Efficacy and Safety of PARP Inhibitors in Metastatic Castration-Resistant Prostate Cancer: A Systematic Review and Meta-Analysis of Clinical Trials. Cancer Treat. Rev. 2023, 120, 102623.
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 1, 1–16.
- Van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The Genomic Landscape of Metastatic Castration-Resistant Prostate Cancers Reveals Multiple Distinct Genotypes with Potential Clinical Impact. Nat. Commun. 2019, 10, 5251.
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 175, 889.
- Saraon, P.; Jarvi, K.; Diamandis, E.P. Molecular Alterations during Progression of Prostate Cancer to Androgen Independence. Clin. Chem. 2011, 57, 1366–1375.
- Powell, S.M.; Christiaens, V.; Voulgaraki, D.; Waxman, J.; Claessens, F.; Bevan, C.L. Mechanisms of Androgen Receptor Signalling via Steroid Receptor Coactivator-1 in Prostate. Endocr. Relat. Cancer 2004, 11, 117–130.
- Taplin, M.-E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the Androgen-Receptor Gene in Metastatic Androgen-Independent Prostate Cancer. N. Engl. J. Med. 1995, 332, 1393–1398.
- Newmark, J.R.; Hardy, D.O.; Tonb, D.C.; Carter, B.S.; Epstein, J.I.; Isaacs, W.B.; Brown, T.R.; Barrack, E.R. Androgen Receptor Gene Mutations in Human Prostate Cancer. Proc. Natl. Acad. Sci. USA 1992, 89, 6319–6323.
- Wyatt, A.W.; Gleave, M.E. Targeting the Adaptive Molecular Landscape of Castration-Resistant Prostate Cancer. EMBO Mol. Med. 2015, 7, 878–894.
- Wilding, G.; Chen, M.; Gelmann, E.P. Aberrant Response in Vitro of Hormone-Responsive Prostate Cancer Cells to Antiandrogens. Prostate 1989, 14, 103–115.
- Taplin, M.-E.; Bubley, G.J.; Ko, Y.-J.; Small, E.J.; Upton, M.; Rajeshkumar, B.; Balk, S.P. Selection for Androgen Receptor Mutations in Prostate Cancers Treated with Androgen Antagonist1. Cancer Res. 1999, 59, 2511–2515.
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen Receptor Activation in Prostatic Tumor Cell Lines by Insulin-Like Growth Factor-1,Keratinocyte Growth Factor and Epidermal Growth Factor. Eur. Urol. 2017, 27, 45–47.
- Hara, T.; Miyazaki, J.; Araki, H.; Yamaoka, M.; Kanzaki, N.; Kusaka, M.; Miyamoto, M. Novel Mutations of Androgen Receptor: A Possible Mechanism of Bicalutamide Withdrawal Syndrome. Cancer Res. 2003, 63, 149–153.
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging Mechanisms of Resistance to Androgen Receptor Inhibitors in Prostate Cancer. Nat. Rev. Cancer 2015, 15, 701–711.
- Tan, J.; Sharief, Y.; Hamil, K.G.; Gregory, C.W.; Zang, D.Y.; Sar, M.; Gumerlock, P.H.; White, R.W.D.; Pretlow, T.G.; Harris, S.E.; et al. Dehydroepiandrosterone Activates Mutant Androgen Receptors Expressed in the Androgen-Dependent Human Prostate Cancer Xenograft CWR22 and LNCaP Cells. Mol. Endocrinol. 1997, 11, 450–459.
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A Novel Androgen Receptor Splice Variant Is Up-Regulated during Prostate Cancer Progression and Promotes Androgen Depletion–Resistant Growth. Cancer Res. 2009, 69, 2305–2313.
- Zhang, T.; Karsh, L.I.; Nissenblatt, M.J.; Canfield, S.E. Androgen Receptor Splice Variant, AR-V7, as a Biomarker of Resistance to Androgen Axis-Targeted Therapies in Advanced Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, 1–10.
- Saraon, P.; Drabovich, A.P.; Jarvi, K.A.; Diamandis, E.P. Mechanisms of Androgen-Independent Prostate Cancer. EJIFCC 2014, 25, 42–54.
- Heinlein, C.A.; Chang, C. Androgen Receptor (AR) Coregulators: An Overview. Endocr. Rev. 2002, 23, 175–200.
- Gregory, C.W.; He, B.; Johnson, R.T.; Ford, O.H.; Mohler, J.L.; French, F.S.; Wilson, E.M. A Mechanism for Androgen Receptor-Mediated Prostate Cancer Recurrence after Androgen Deprivation Therapy. Cancer Res. 2001, 61, 4315–4319.
- Zhou, X.E.; Suino-Powell, K.M.; Li, J.; He, Y.; Mackeigan, J.P.; Melcher, K.; Yong, E.-L.; Xu, H.E. Identification of SRC3/AIB1 as a Preferred Coactivator for Hormone-Activated Androgen Receptor. J. Biol. Chem. 2010, 285, 9161–9171.
- Tien, J.C.-Y.; Liu, Z.; Liao, L.; Wang, F.; Xu, Y.; Wu, Y.-L.; Zhou, N.; Ittmann, M.; Xu, J. The Steroid Receptor Coactivator-3 Is Required for the Development of Castration-Resistant Prostate Cancer. Cancer Res. 2013, 73, 3997–4008.
- Cheng, S.; Brzostek, S.; Lee, S.R.; Hollenberg, A.N.; Balk, S.P. Inhibition of the Dihydrotestosterone-Activated Androgen Receptor by Nuclear Receptor Corepressor. Mol. Endocrinol. 2002, 16, 1492–1501.
- Godoy, A.S.; Sotomayor, P.C.; Villagran, M.; Yacoub, R.; Montecinos, V.P.; McNerney, E.M.; Moser, M.; Foster, B.A.; Onate, S.A. Altered Corepressor SMRT Expression and Recruitment to Target Genes as a Mechanism That Change the Response to Androgens in Prostate Cancer Progression. Biochem. Biophys. Res. Commun. 2012, 423, 564–570.
- Liao, G.; Chen, L.-Y.; Zhang, A.; Godavarthy, A.; Xia, F.; Ghosh, J.C.; Li, H.; Chen, J.D. Regulation of Androgen Receptor Activity by the Nuclear Receptor Corepressor SMRT*. J. Biol. Chem. 2003, 278, 5052–5061.
- Nacusi, L.P.; Tindall, D.J. Targeting 5α-Reductase for Prostate Cancer Prevention and Treatment. Nat. Rev. Urol. 2011, 8, 378–384.
- Dutt, S.S.; Gao, A.C. Molecular Mechanisms of Castration-Resistant Prostate Cancer Progression. Future Oncol. 2009, 5, 1403–1413.
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of Intratumoral Androgens in Metastatic Prostate Cancer: A Mechanism for Castration-Resistant Tumor Growth. Cancer Res. 2008, 68, 4447–4454.
- Wu, J.D.; Haugk, K.; Woodke, L.; Nelson, P.; Coleman, I.; Plymate, S.R. Interaction of IGF Signaling and the Androgen Receptor in Prostate Cancer Progression. J. Cell. Biochem. 2006, 99, 392–401.
- Malinowska, K.; Neuwirt, H.; Cavarretta, I.T.; Bektic, J.; Steiner, H.; Dietrich, H.; Moser, P.L.; Fuchs, D.; Hobisch, A.; Culig, Z. Interleukin-6 Stimulation of Growth of Prostate Cancer in Vitro and in Vivo through Activation of the Androgen Receptor. Endocr. Relat. Cancer 2009, 16, 155–169.
- Sarker, D.; Reid, A.H.M.; Yap, T.A.; de Bono, J.S. Targeting the PI3K/AKT Pathway for the Treatment of Prostate Cancer. Clin. Cancer Res. 2009, 15, 4799–4805.
- Wen, Y.; Hu, M.C.; Makino, K.; Spohn, B.; Bartholomeusz, G.; Yan, D.H.; Hung, M.C. HER-2/Neu Promotes Androgen-Independent Survival and Growth of Prostate Cancer Cells through the Akt Pathway. Cancer Res. 2000, 60, 6841–6845.
- Graff, J.R.; Konicek, B.W.; McNulty, A.M.; Wang, Z.; Houck, K.; Allen, S.; Paul, J.D.; Hbaiu, A.; Goode, R.G.; Sandusky, G.E.; et al. Increased AKT Activity Contributes to Prostate Cancer Progression by Dramatically Accelerating Prostate Tumor Growth and Diminishing p27Kip1 Expression. J. Biol. Chem. 2000, 275, 24500–24505.
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical Implications of PTEN Loss in Prostate Cancer. Nat. Rev. Urol. 2018, 15, 222–234.
- Andrieu, C.; Taieb, D.; Baylot, V.; Ettinger, S.; Soubeyran, P.; De-Thonel, A.; Nelson, C.; Garrido, C.; So, A.; Fazli, L.; et al. Heat Shock Protein 27 Confers Resistance to Androgen Ablation and Chemotherapy in Prostate Cancer Cells through eIF4E. Oncogene 2010, 29, 1883–1896.
- Baylot, V.; Katsogiannou, M.; Andrieu, C.; Taieb, D.; Acunzo, J.; Giusiano, S.; Fazli, L.; Gleave, M.; Garrido, C.; Rocchi, P. Targeting TCTP as a New Therapeutic Strategy in Castration-Resistant Prostate Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 2244–2256.
- Cherif, C.; Nguyen, D.T.; Paris, C.; Le, T.K.; Sefiane, T.; Carbuccia, N.; Finetti, P.; Chaffanet, M.; Kaoutari, A.E.; Vernerey, J.; et al. Menin Inhibition Suppresses Castration-Resistant Prostate Cancer and Enhances Chemosensitivity. Oncogene 2022, 41, 125–137.
- Le, T.K.; Cherif, C.; Omabe, K.; Paris, C.; Lannes, F.; Audebert, S.; Baudelet, E.; Hamimed, M.; Barbolosi, D.; Finetti, P.; et al. DDX5 mRNA-Targeting Antisense Oligonucleotide as a New Promising Therapeutic in Combating Castration-Resistant Prostate Cancer. Mol. Ther. 2022, 31, 471–486.
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell 2013, 155, 1309–1322.
- Puhr, M.; Hoefer, J.; Eigentler, A.; Ploner, C.; Handle, F.; Schaefer, G.; Kroon, J.; Leo, A.; Heidegger, I.; Eder, I.; et al. The Glucocorticoid Receptor Is a Key Player for Prostate Cancer Cell Survival and a Target for Improved Antiandrogen Therapy. Clin. Cancer Res. 2018, 24, 927–938.
- Xie, N.; Cheng, H.; Lin, D.; Liu, L.; Yang, O.; Jia, L.; Fazli, L.; Gleave, M.E.; Wang, Y.; Rennie, P.; et al. The Expression of Glucocorticoid Receptor Is Negatively Regulated by Active Androgen Receptor Signaling in Prostate Tumors. Int. J. Cancer 2015, 136, E27–E38.
- Patel, G.K.; Chugh, N.; Tripathi, M. Neuroendocrine Differentiation of Prostate Cancer—An Intriguing Example of Tumor Evolution at Play. Cancers 2019, 11, 1405.
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive Variants of Castration Resistant Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2846–2850.
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discov. 2011, 1, 487–495.
- Lin, D.; Wyatt, A.W.; Xue, H.; Wang, Y.; Dong, X.; Haegert, A.; Wu, R.; Brahmbhatt, S.; Mo, F.; Jong, L.; et al. High Fidelity Patient-Derived Xenografts for Accelerating Prostate Cancer Discovery and Drug Development. Cancer Res. 2014, 74, 1272–1283.
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 Cooperate to Suppress Prostate Cancer Lineage Plasticity, Metastasis, and Antiandrogen Resistance. Science 2017, 355, 78–83.
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017, 7, 736–749.
- Deng, X.; Liu, H.; Huang, J.; Cheng, L.; Keller, E.T.; Parsons, S.J.; Hu, C.-D. Ionizing Radiation Induces Prostate Cancer Neuroendocrine Differentiation through Interplay of CREB and ATF2: Implications for Disease Progression. Cancer Res. 2008, 68, 9663–9670.
- Bonkhoff, H. Factors Implicated in Radiation Therapy Failure and Radiosensitization of Prostate Cancer. Prostate Cancer 2012, 2012, 593241.
- Berruti, A.; Dogliotti, L.; Mosca, A.; Bellina, M.; Mari, M.; Torta, M.; Tarabuzzi, R.; Bollito, E.; Fontana, D.; Angeli, A. Circulating Neuroendocrine Markers in Patients with Prostate Carcinoma. Cancer 2000, 88, 2590–2597.
- Davies, A.; Zoubeidi, A.; Selth, L.A. The Epigenetic and Transcriptional Landscape of Neuroendocrine Prostate Cancer. Endocr. Relat. Cancer 2020, 27, R35–R50.
- Baca, S.C.; Takeda, D.Y.; Seo, J.-H.; Hwang, J.; Ku, S.Y.; Arafeh, R.; Arnoff, T.; Agarwal, S.; Bell, C.; O’Connor, E.; et al. Reprogramming of the FOXA1 Cistrome in Treatment-Emergent Neuroendocrine Prostate Cancer. Nat. Commun. 2021, 12, 1979.
- Dang, Q.; Li, L.; Xie, H.; He, D.; Chen, J.; Song, W.; Chang, L.S.; Chang, H.-C.; Yeh, S.; Chang, C. Anti-Androgen Enzalutamide Enhances Prostate Cancer Neuroendocrine (NE) Differentiation via Altering the Infiltrated Mast Cells→Androgen Receptor (AR)→miRNA32 Signals. Mol. Oncol. 2015, 9, 1241–1251.
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Agarwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal Epigenetic Alterations Drive Metabolic and Neuroendocrine Prostate Cancer Reprogramming. J. Clin. Investig. 2018, 128, 4472–4484.


