Prostate cancer (PC) is the second most common cancer in men worldwide. Despite recent advances in diagnosis and treatment, castration-resistant prostate cancer (CRPC) remains a significant medical challenge. Prostate cancer cells can develop mechanisms to resist androgen deprivation therapy, such as AR overexpression, AR mutations, alterations in AR coregulators, increased steroidogenic signaling pathways, outlaw pathways, and bypass pathways. Various treatment options for CRPC exist, including androgen deprivation therapy, chemotherapy, immunotherapy, localized or systemic therapeutic radiation, and PARP inhibitors. However, more research is needed to combat CRPC effectively. Further investigation into the underlying mechanisms of the disease and the development of new therapeutic strategies will be crucial in improving patient outcomes.
1. Introduction
Prostate cancer (PC) is the second most prevalent cancer in men worldwide, representing 15% of all cancers diagnosed in males
[1]. In the US, it is the most frequently diagnosed cancer and the second leading cause of cancer deaths in men. An estimated 288,300 new PC cases and 34,700 deaths from the disease are predicted to occur in the US in 2023
[2]. Patients who have advanced PC initially respond very well to androgen deprivation therapy. However, the treatment finally selects cancer cells that relapse into androgen deprivation, leading to the emergence of castration-resistant prostate cancer (CRPC). Patients with metastatic CRPC (mCRPC) have reduced life expectancy, with a median overall survival rate of below 2 years. The current definition CRPC, according to the European Association of Urology (EAU), is based on biochemical and/or clinical parameters demonstrating cancer progression in a castration environment (testosterone level < 50 ng/dL or 1.7 nmol/L) and standards proposed by Prostate Cancer Working Group 3 (PCWG3)
[3] and/or the Response Evaluation Criteria in Solid Tumors (RECIST 1.1)
[4]. Accordingly, biological progression refers to a condition where there are three consecutive increases in PSA (Prostate Specific Antigen), each at least one week apart, resulting in two increases of at least 50% over the lowest point, and a PSA level greater than 2 ng/mL. Radiographic progression is defined by the appearance of at least two new lesions on a bone scan or the progression of a measurable lesion according to the RECIST
[5].
Over the past decade, there has been significant improvement in the treatment options for CRPC. Several drugs that increase overall patient survival, including androgen-receptor (AR) signaling inhibitors (such as abiraterone acetate, enzalutamide, apalutamide, and darolutamide), and radiopharmaceutical therapies (like radium-223 and 177Lu-PSMA-617) have been approved
[6]. Advancements in precision medicine have led to the discovery of distinct subtypes of prostate cancer and the identification of genetic changes that predict the effectiveness of specific treatments
[7][8][7,8].
2. Mechanisms Underlying Castration-Resistant Prostate Cancer
There are currently many treatment options available for CRPC patients; however, the responses to these therapies are still limited. Knowledge of the underlying mechanisms associated with the CRPC phenotype can help to identify new promising therapeutics.
2.1. Androgen Receptor Overexpression
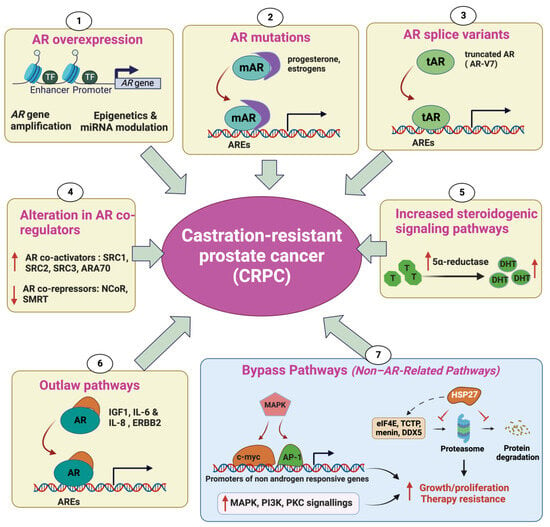
Androgen Receptor (AR) overexpression enables the survival and proliferation of tumor cells in limited-androgen conditions during androgen suppression treatment. Elevated AR expression at both the mRNA and protein levels has been frequently observed in CRPC. Amplification of the AR has been observed in 70% of cases of CRPC and is associated with significantly increased AR mRNA expression
[9]. David A. et al. reported the existence of an enhancer that is amplified in 81% of castration-resistant metastatic patients. This enhancer has the potential to increase the expression of the
AR gene independently of AR locus amplification in response to first-line androgen deprivation therapy (ADT)
[9]. It should be noted that AR amplification is only seen in prostate tumors that have been exposed to androgen deprivation, indicating that AR amplification is a consequence of hormone therapy
[10]. Additionally, epigenetic modifications, such as DNA methylation, histone acetylation, and miRNA modulation, could lead to AR overexpression in CRPC (
Figure 1)
[11].
Figure 1. Mechanisms of castration-resistant prostate cancer (CRPC). AR overexpression enables the survival and proliferation of tumor cells in limited-androgen conditions during androgen suppression treatment. A high AR expression level can be due to AR gene amplification, epigenetics, and miRNA modulation (1). Point mutations in the ligand-binding domain of the AR gene lead to the increased affinity of the mutated AR (mAR) to other hormones, such as progesterone and estrogen, thereby modulating androgen-responsive gene transcription independently from androgen (2). The emergence of AR-splicing isoforms, such as AR3 (also called AR-V7), AR4, and AR5, encoding the truncated AR protein (tAR), which lacks the ligand-binding domain, results in constitutive activation of the AR, thereby promoting variant-carrying tumor cells to ignore the need for androgen (3). Overexpression of AR coactivators and the decreased expression of AR corepressors will result in an increase in AR-regulated transcription (4). Increased production of 5α-reductase can provide sufficient androgens for AR activation in cancer cells (5). CRPC can be induced by outlaw pathways in which AR signaling can be activated in a ligand-independent manner by other molecules than androgens, such as growth factors, cytokines, and kinases (6). The bypass pathways, involved in CRPC progression, increase the activity of MAPK, PI3K, and PCK cascades, leading to either the stimulation of alternative growth pathways or the enhancement of survival signaling independently from AR signaling. Alternative pathways also involve the overexpression of heat-shock protein 27 (HSP27), which mediates its cytoprotective function by protecting its interacting proteins (eIF4E, TCTP, Menin, DDX5) from their degradation by the proteasome (7) (↑ in red: increase, ↓in red: decrease).
2.2. AR Mutations
AR gene mutations have been found in around 10–20% of CRPC cases, while they are rarely observed in localized PC (0–4%)
[12][13][12,13]. Most recurrent AR mutations are found in the ligand-binding domain (LBD) and/or cofactor binding regions
[14]. These alterations could decrease the specificity of the AR for its main ligand, androgen; however, they allow AR specificity to extend to other hormones, such as progesterone and estrogen
[12][15][16][17][18][12,15,16,17,18]. In addition, they could also disrupt the effectiveness of AR antagonists, causing them to act as AR agonists and resulting in resistance to treatments
[19]. It has been reported that the H875Y mutation in CWR22 causes cells to exhibit altered ligand specificity. This mutant AR has shown transcriptional activity in response to testicular androgens, similarly to the wild-type AR. However, it differs from the wild type in its additional activation by adrenal androgens, dehydroepiandrosterone, and the antiandrogen hydroxyflutamide, as well as by estradiol and progesterone
[20]. Along with AR coding mutations, several AR splicing mutations, leading to the abnormal expression of splice variants such as AR3 (also called AR-V7), AR4, and AR5, which lack the ligand-binding domain, are frequently observed in CRPC
[21]. These variants encode a shorter receptor that lacks the ligand-binding domain and thereby constitutively activates AR pathways
[22].
2.3. Alteration in AR CoRegulators
As a transcription factor (TF), the AR cooperates with a variety of regulatory proteins to form a productive transcription complex. These coregulators can either enhance (coactivators) or depress (cosuppressors) the transcriptional activity of the AR, thereby modulating the expression of androgen-regulated genes. As a consequence, alteration in the expression of AR coregulators can give a survival advantage to the cancer cells during castration therapy, promoting CRPC progression
[23]. Several well-documented AR coactivators, including SRC1, SRC2, SRC3, ARA70, PIAS1, and Tip60, interact with the AR and enhance its transcriptional activity
[24]. Levels of TIF2 and SRC1 expression have been observed to rise alongside the expression of the AR during the progression of prostate cancer cells following androgen deprivation. A previous study has also shown that these proteins are overexpressed in the majority of CRPC samples. The overexpression of TIF2 enhances the transcriptional activation of the AR in response to weaker steroids, such as adrenal androgens (DHEA and androstenedione), in two cell lines carrying AR mutations: LNCaP (T877A) and CWR22 (H874Y)
[25]. The steroid receptor coactivator-3 (SRC3), also called amplified-in-breast cancer-1 (AIB1), has been identified as a potent coactivator of the hormone-activated AR
[26] and has been demonstrated to promote CRPC progression
[27]. Conversely, two well-known corepressors of the AR, NCoR (nuclear receptor corepressor) and SMRT (silencing mediator for retinoid and thyroid hormone receptors), have been shown to interact with the AR and decrease its transcriptional activity triggered by dihydrotestosterone
[28][29][30][28,29,30].
2.4. Increased Steroidogenic Signaling Pathways
Accumulating evidence has reported that PC cells could survive after castration therapy by regulating intracrine androgen synthesis within the prostate. A typical example is the 5α-reductase enzyme, which catalyzes the conversion of testosterone (the most abundant free androgen) to the higher-affinity ligand 5α-dihydrotestosterone (DHT). Upon ADT, intraprostatic testosterone and DHT levels do not decline as markedly as do serum levels after ADT, buffering tumor cells from the loss of testicular androgen
[31]. Increased 5α-reductase enzyme levels result from a polymorphism where there is a substitution of a valine with a leucine at the codon 89
[32]. Intraprostatic androgens can also be synthesized from cholesterol or other molecular precursors, such as DHEA (dehydroepiandrosterone). Indeed, DHEA could be converted into androstenedione, a substrate for conversion to testosterone. Another report has shown that CRPC samples have higher levels of expression of a variety of enzymes involved in de novo steroid synthesis, such as FASN, CYP17A1, CYP19A1, and UGT2B17, compared to primary PC
[33].
2.5. Outlaw Pathways
In addition of being activated primarily by endogenous androgen ligands, the AR protein can also be stimulated through ligand-independent mechanisms, which are known as outlaw pathways. CRPC can thus be induced by the interactions between the cytosolic AR and many molecules such as different growth factors, cytokines, and kinases. Several growth factors, such as insulin-like growth factor-I (IGF-I), keratinocyte growth factor (KGF) and the epidermal growth factor (EGF), enable the stimulation of AR transcription activity at limited androgen levels or even in the absence of androgens
[17]. Particularly, IGF1 has been reported to interact with the AR, facilitate AR translocation, and modulate the transcription of androgen-responsive genes. Moreover, it has been demonstrated to upregulate the expression of the AR coactivator TIF2
[34]. Along with growth factors, many cytokines, such as IL-6 and IL-8, have proven their ability to activate AR signaling in a ligand-independent manner
[35]. Additionally, receptor tyrosine kinases (RTK) such as HER2/ERBB2 have been shown to be overexpressed in CRPC and restore AR signaling in low-androgen-concentration conditions
[25].
2.6. Non-AR-Related Pathways: Bypass Pathways
While the mechanisms described above rely on AR transactivation to promote CRPC progression, alternative survival and growth pathways that are independent of AR activation can also drive androgen deprivation resistance. Of note, many outlaw pathways can also drive CRPC progression in an AR-signaling independent manner. High levels of serum insulin-like growth factor-I (IGF-I) and its receptor (IGF-1R) have been increasingly recognized to play a key role in prostate cancer progression. Binding of IGF-I with IGF-IR enables the stimulation of the transduction of a signal cascade, which, in turn, activates the expression of genes responsible for cellular growth and proliferation
[10][23][10,23]. The upregulation of Akt pathway has been reported in a variety of human malignancies, including prostate cancer
[36]. Indeed, AKT plays a central role in antiapoptotic pathways by phosphorylating and depressing Bcl2-associated agonist of cell death (BAD), a proapoptotic member of the BCL-2 protein family, and pro-caspase 9
[37]. AKT also promotes cellular proliferation by decreasing the expression of p27, a cell cycle inhibitor
[38]. Several reports have shown that loss of PTEN, a tumor suppressor that inhibits PI3K/AKT signaling, frequently occurs in CRPC samples
[39].
In addition, the heat-shock protein Hsp27 has previously been demonstrated to drive therapy resistance in PC, and its inhibitors, such as Hsp27 ASO (OGX-427) and small interference RNA (siRNA), have shown the ability to enhance chemotherapy. More recently, people have shown that Hsp27 plays a role in promoting the development of CRPC through its chaperone activity by selectively protecting some partner proteins involved in castration resistance, such as eIF4E, TCTP, Menin, and DDX5, from their ubiquitin–proteasome degradation
[40][41][42][43][40,41,42,43]. CRPC has the potential to develop resistance to the antiandrogen enzalutamide by bypassing the AR blockade, which occurs due to increased activity of the glucocorticoid receptor (GR)
[44]. Recent studies have indicated that heightened activity of the GR has been linked to the emergence of resistance to antiandrogen therapies in different clinical and preclinical models
[44][45][44,45]. These findings could possibly be explained by the fact that GR expression is downregulated by AR signaling
[46]. Additionally, the GR interacts with and mediates a subset of AR targets
[44].
2.7. Evolution of t-NEPC from CRPC
During the initial diagnosis of PC, neuroendocrine prostate cancer (NEPC) is rare and represents only 0.5–2% of all PC cases. However, a larger proportion of treatment-induced neuroendocrine prostate cancer (t-NEPC) has been reported. Recent studies have indicated that approximately 17–30% of CRPC patients, following ADT and other treatments, may experience progression to t-NEPC
[47][48][47,48]. The majority of evidence suggests that the origin of tNEPC is the transdifferentiation of adenocarcinoma cells into NEPC cells in response to different therapies, including ADT
[47][48][49][47,48,49]. The transition to the NEPC phenotype following ADT treatments has been demonstrated in various preclinical models, including cell lines, genetically engineered mouse (GEM), and patient-derived xenografts
[50][51][52][50,51,52]. In addition to ADT, other therapies, such as radiation and chemotherapy, have been reported to stimulate the NEPC phenotype
[53][54][55][53,54,55]. Moreover, genetic and epigenetic alterations and tumor microenvironments (TMEs) containing a variety of cells and factors could support NEPC transition in CRPC patients
[56][57][58][59][56,57,58,59]. Due to the absence of AR and PSA expression (AR-/PSA-), t-NEPC patients have shown limited responses to existing treatment options. To date, several therapeutics have been proposed for patients with t-NEPC, including cisplatin or carboplatin combined with etoposide or docetaxel.