 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Amaresh Ranjan | -- | 4870 | 2023-10-27 23:49:40 | | | |
| 2 | Lindsay Dong | Meta information modification | 4870 | 2023-10-30 02:07:08 | | |
Video Upload Options
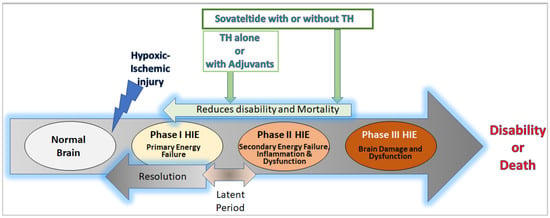
Neonatal hypoxic-ischemic encephalopathy (HIE) is a condition that results in brain damage in newborns due to insufficient blood and oxygen supply during or after birth. HIE is a major cause of neurological disability and mortality in newborns, with over one million neonatal deaths occurring annually worldwide. The severity of brain injury and the outcome of HIE depend on several factors, including the cause of oxygen deprivation, brain maturity, regional blood flow, and maternal health conditions. HIE is classified into mild, moderate, and severe categories based on the extent of brain damage and resulting neurological issues. The pathophysiology of HIE involves different phases, including the primary phase, latent phase, secondary phase, and tertiary phase. The primary and secondary phases are characterized by episodes of energy and cell metabolism failures, increased cytotoxicity and apoptosis, and activated microglia and inflammation in the brain.
1. Introduction
2. HIE Pathology, Pathophysiology, and Neural Cell Damage
2.1. Pathology
2.2. Pathophysiology and Neural Cell Death
3. Current Interventions or Therapies for Neonatal HIE
Therapeutic Hypothermia
4. Therapies under Development for Neonatal HIE
4.1. Therapies under Development with Clinical Study Results
4.1.1. Xenon
4.1.2. Erythropoietin and Darbepoetin
4.1.3. Topiramate
4.1.4. Glucocorticoids
4.2. Future Therapies (Therapies under Development with Results Awaited)
4.2.1. Melatonin
4.2.2. Caffeine
4.2.3. Citicoline
4.2.4. Metformin
4.2.5. Allopurinol
4.2.6. RLS 0071
4.2.7. Stem Cells
4.2.8. Sovateltide
5. Conclusions
References
- Lawn, J.E.; Cousens, S.; Zupan, J.; Lancet Neonatal Survival Steering Team. 4 million neonatal deaths: When? Where? Why? Lancet 2005, 365, 891–900.
- Allen, K.A.; Brandon, D.H. Hypoxic Ischemic Encephalopathy: Pathophysiology and Experimental Treatments. Newborn Infant Nurs. Rev. 2011, 11, 125–133.
- Bruschettini, M.; Romantsik, O.; Moreira, A.; Ley, D.; Thébaud, B. Stem cell-based interventions for the prevention of morbidity and mortality following hypoxic-ischaemic encephalopathy in newborn infants. Cochrane Database Syst. Rev. 2020, 8, Cd013202.
- Andersen, M.; Andelius, T.C.K.; Pedersen, M.V.; Kyng, K.J.; Henriksen, T.B. Severity of hypoxic ischemic encephalopathy and heart rate variability in neonates: A systematic review. BMC Pediatr. 2019, 19, 242.
- Yokomaku, D.; Numakawa, T.; Numakawa, Y.; Suzuki, S.; Matsumoto, T.; Adachi, N.; Nishio, C.; Taguchi, T.; Hatanaka, H. Estrogen enhances depolarization-induced glutamate release through activation of phosphatidylinositol 3-kinase and mitogen-activated protein kinase in cultured hippocampal neurons. Mol. Endocrinol. 2003, 17, 831–844.
- Fatemi, A.; Wilson, M.A.; Johnston, M.V. Hypoxic-ischemic encephalopathy in the term infant. Clin. Perinatol. 2009, 36, 835–858, vii.
- Nair, J.; Kumar, V.H.S. Current and Emerging Therapies in the Management of Hypoxic Ischemic Encephalopathy in Neonates. Children 2018, 5, 99.
- Riljak, V.; Kraf, J.; Daryanani, A.; Jiruska, P.; Otahal, J. Pathophysiology of perinatal hypoxic-ischemic encephalopathy—Biomarkers, animal models and treatment perspectives. Physiol. Res. 2016, 65, S533–S545.
- Gagne-Loranger, M.; Sheppard, M.; Ali, N.; Saint-Martin, C.; Wintermark, P. Newborns Referred for Therapeutic Hypothermia: Association between Initial Degree of Encephalopathy and Severity of Brain Injury (What about the Newborns with Mild Encephalopathy on Admission?). Am. J. Perinatol. 2016, 33, 195–202.
- Walsh, B.H.; Neil, J.; Morey, J.; Yang, E.; Silvera, M.V.; Inder, T.E.; Ortinau, C. The Frequency and Severity of Magnetic Resonance Imaging Abnormalities in Infants with Mild Neonatal Encephalopathy. J. Pediatr. 2017, 187, 26–33.e21.
- Rao, R.; Trivedi, S.; Distler, A.; Liao, S.; Vesoulis, Z.; Smyser, C.; Mathur, A.M. Neurodevelopmental Outcomes in Neonates with Mild Hypoxic Ischemic Encephalopathy Treated with Therapeutic Hypothermia. Am. J. Perinatol. 2019, 36, 1337–1343.
- Massaro, A.N.; Murthy, K.; Zaniletti, I.; Cook, N.; DiGeronimo, R.; Dizon, M.; Hamrick, S.E.; McKay, V.J.; Natarajan, G.; Rao, R.; et al. Short-term outcomes after perinatal hypoxic ischemic encephalopathy: A report from the Children’s Hospitals Neonatal Consortium HIE focus group. J. Perinatol. 2015, 35, 290–296.
- Conway, J.M.; Walsh, B.H.; Boylan, G.B.; Murray, D.M. Mild hypoxic ischaemic encephalopathy and long term neurodevelopmental outcome—A systematic review. Early Hum. Dev. 2018, 120, 80–87.
- Pandav, K.; Ishak, A.; Chohan, F.; Edaki, O.; Quinonez, J.; Ruxmohan, S. Hypoxic-Ischemic Encephalopathy-Induced Seizure in an 11-Year-Old Female. Cureus 2021, 13, e16606.
- Northington, F.J.; Chavez-Valdez, R.; Martin, L.J. Neuronal cell death in neonatal hypoxia-ischemia. Ann. Neurol. 2011, 69, 743–758.
- Dickey, E.J.; Long, S.N.; Hunt, R.W. Hypoxic ischemic encephalopathy—What can we learn from humans? J. Vet. Intern. Med. 2011, 25, 1231–1240.
- Zhao, M.; Zhu, P.; Fujino, M.; Zhuang, J.; Guo, H.; Sheikh, I.; Zhao, L.; Li, X.K. Oxidative Stress in Hypoxic-Ischemic Encephalopathy: Molecular Mechanisms and Therapeutic Strategies. Int. J. Mol. Sci. 2016, 17, 2078.
- Ozsurekci, Y.; Aykac, K. Oxidative Stress Related Diseases in Newborns. Oxid. Med. Cell. Longev. 2016, 2016, 2768365.
- Shankaran, S. Neonatal encephalopathy: Treatment with hypothermia. J. Neurotrauma 2009, 26, 437–443.
- Weilinger, N.L.; Maslieieva, V.; Bialecki, J.; Sridharan, S.S.; Tang, P.L.; Thompson, R.J. Ionotropic receptors and ion channels in ischemic neuronal death and dysfunction. Acta Pharmacol. Sin. 2013, 34, 39–48.
- Stys, P.K. Anoxic and ischemic injury of myelinated axons in CNS white matter: From mechanistic concepts to therapeutics. J. Cereb. Blood Flow Metab. 1998, 18, 2–25.
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317.
- Ranneh, Y.; Ali, F.; Akim, A.M.; Hamid, H.A.; Khazaai, H.; Fadel, A. Crosstalk between reactive oxygen species and pro-inflammatory markers in developing various chronic diseases: A review. Appl. Biol. Chem. 2017, 60, 327–338.
- Mallard, C.; Tremblay, M.E.; Vexler, Z.S. Microglia and Neonatal Brain Injury. Neuroscience 2019, 405, 68–76.
- Liu, F.; McCullough, L.D. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol. Sin. 2013, 34, 1121–1130.
- Min, Y.J.; Ling, E.A.; Li, F. Immunomodulatory Mechanism and Potential Therapies for Perinatal Hypoxic-Ischemic Brain Damage. Front. Pharmacol. 2020, 11, 580428.
- Rayasam, A.; Fukuzaki, Y.; Vexler, Z.S. Microglia-leucocyte axis in cerebral ischaemia and inflammation in the developing brain. Acta Physiol. 2021, 233, e13674.
- Jha, M.K.; Jeon, S.; Suk, K. Glia as a Link between Neuroinflammation and Neuropathic Pain. Immune Netw. 2012, 12, 41–47.
- Leavy, A.; Jimenez Mateos, E.M. Perinatal Brain Injury and Inflammation: Lessons from Experimental Murine Models. Cells 2020, 9, 2640.
- Li, B.; Concepcion, K.; Meng, X.; Zhang, L. Brain-immune interactions in perinatal hypoxic-ischemic brain injury. Prog. Neurobiol. 2017, 159, 50–68.
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215.
- Peliowski-Davidovich, A.; Canadian Paediatric Society, F.; Newborn, C. Hypothermia for newborns with hypoxic ischemic encephalopathy. Paediatr. Child Health 2012, 17, 41–46.
- Wassink, G.; Gunn, E.R.; Drury, P.P.; Bennet, L.; Gunn, A.J. The mechanisms and treatment of asphyxial encephalopathy. Front. Neurosci. 2014, 8, 40.
- Cornette, L. Therapeutic hypothermia in neonatal asphyxia. Facts Views Vis. ObGyn 2012, 4, 133–139.
- Whitelaw, A.; Thoresen, M. Therapeutic Hypothermia for Hypoxic-Ischemic Brain Injury Is More Effective in Newborn Infants than in Older Patients: Review and Hypotheses. Ther. Hypothermia Temp. Manag. 2023; ahead of print.
- Kurisu, K.; Kim, J.Y.; You, J.; Yenari, M.A. Therapeutic Hypothermia and Neuroprotection in Acute Neurological Disease. Curr. Med. Chem. 2019, 26, 5430–5455.
- Lemyre, B.; Chau, V. Hypothermia for newborns with hypoxic-ischemic encephalopathy. Paediatr. Child Health 2018, 23, 285–291.
- Kendall, G.S.; Mathieson, S.; Meek, J.; Rennie, J.M. Recooling for rebound seizures after rewarming in neonatal encephalopathy. Pediatrics 2012, 130, e451–e455.
- Karcioglu, O.; Topacoglu, H.; Dikme, O.; Dikme, O. A systematic review of safety and adverse effects in the practice of therapeutic hypothermia. Am. J. Emerg. Med. 2018, 36, 1886–1894.
- Simons, S.H.; van Dijk, M.; van Lingen, R.A.; Roofthooft, D.; Boomsma, F.; van den Anker, J.N.; Tibboel, D. Randomised controlled trial evaluating effects of morphine on plasma adrenaline/noradrenaline concentrations in newborns. Arch. Dis. Child. Fetal Neonatal Ed. 2005, 90, F36–F40.
- Frymoyer, A.; Bonifacio, S.L.; Drover, D.R.; Su, F.; Wustoff, C.J.; Van Meurs, K.P. Decreased Morphine Clearance in Neonates with Hypoxic Ischemic Encephalopathy Receiving Hypothermia. J. Clin. Pharmacol. 2017, 57, 64–76.
- Sabir, H.; Maes, E.; Zweyer, M.; Schleehuber, Y.; Imam, F.B.; Silverman, J.; White, Y.; Pang, R.; Pasca, A.M.; Robertson, N.J.; et al. Comparing the efficacy in reducing brain injury of different neuroprotective agents following neonatal hypoxia-ischemia in newborn rats: A multi-drug randomized controlled screening trial. Sci. Rep. 2023, 13, 9467.
- Ruegger, C.M.; Davis, P.G.; Cheong, J.L. Xenon as an adjuvant to therapeutic hypothermia in near-term and term newborns with hypoxic-ischaemic encephalopathy. Cochrane Database Syst. Rev. 2018, 8, CD012753.
- Anna, R.; Rolf, R.; Mark, C. Update of the organoprotective properties of xenon and argon: From bench to beside. Intensive Care Med. Exp. 2020, 8, 11.
- Esencan, E.; Yuksel, S.; Tosun, Y.B.; Robinot, A.; Solaroglu, I.; Zhang, J.H. XENON in medical area: Emphasis on neuroprotection in hypoxia and anesthesia. Med. Gas Res. 2013, 3, 4.
- Franks, N.P.; Dickinson, R.; de Sousa, S.L.; Hall, A.C.; Lieb, W.R. How does xenon produce anaesthesia? Nature 1998, 396, 324.
- Armstrong, S.P.; Banks, P.J.; McKitrick, T.J.; Geldart, C.H.; Edge, C.J.; Babla, R.; Simillis, C.; Franks, N.P.; Dickinson, R. Identification of two mutations (F758W and F758Y) in the N-methyl-D-aspartate receptor glycine-binding site that selectively prevent competitive inhibition by xenon without affecting glycine binding. Anesthesiology 2012, 117, 38–47.
- Dickinson, R.; Peterson, B.K.; Banks, P.; Simillis, C.; Martin, J.C.; Valenzuela, C.A.; Maze, M.; Franks, N.P. Competitive inhibition at the glycine site of the N-methyl-D-aspartate receptor by the anesthetics xenon and isoflurane: Evidence from molecular modeling and electrophysiology. Anesthesiology 2007, 107, 756–767.
- Petzelt, C.; Blom, P.; Schmehl, W.; Muller, J.; Kox, W.J. Prevention of neurotoxicity in hypoxic cortical neurons by the noble gas xenon. Life Sci. 2003, 72, 1909–1918.
- Dingley, J.; Hobbs, C.; Ferguson, J.; Stone, J.; Thoresen, M. Xenon/hypothermia neuroprotection regimes in spontaneously breathing neonatal rats after hypoxic-ischemic insult: The respiratory and sedative effects. Anesth. Analg. 2008, 106, 916–923.
- Hobbs, C.; Thoresen, M.; Tucker, A.; Aquilina, K.; Chakkarapani, E.; Dingley, J. Xenon and hypothermia combine additively, offering long-term functional and histopathologic neuroprotection after neonatal hypoxia/ischemia. Stroke 2008, 39, 1307–1313.
- Ma, D.; Hossain, M.; Chow, A.; Arshad, M.; Battson, R.M.; Sanders, R.D.; Mehmet, H.; Edwards, A.D.; Franks, N.P.; Maze, M. Xenon and hypothermia combine to provide neuroprotection from neonatal asphyxia. Ann. Neurol. 2005, 58, 182–193.
- Thoresen, M.; Hobbs, C.E.; Wood, T.; Chakkarapani, E.; Dingley, J. Cooling combined with immediate or delayed xenon inhalation provides equivalent long-term neuroprotection after neonatal hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2009, 29, 707–714.
- Chakkarapani, E.; Dingley, J.; Liu, X.; Hoque, N.; Aquilina, K.; Porter, H.; Thoresen, M. Xenon enhances hypothermic neuroprotection in asphyxiated newborn pigs. Ann. Neurol. 2010, 68, 330–341.
- Faulkner, S.; Bainbridge, A.; Kato, T.; Chandrasekaran, M.; Kapetanakis, A.B.; Hristova, M.; Liu, M.; Evans, S.; De Vita, E.; Kelen, D.; et al. Xenon augmented hypothermia reduces early lactate/N-acetylaspartate and cell death in perinatal asphyxia. Ann. Neurol. 2011, 70, 133–150.
- Bantel, C.; Maze, M.; Trapp, S. Noble gas xenon is a novel adenosine triphosphate-sensitive potassium channel opener. Anesthesiology 2010, 112, 623–630.
- Gruss, M.; Bushell, T.J.; Bright, D.P.; Lieb, W.R.; Mathie, A.; Franks, N.P. Two-pore-domain K+ channels are a novel target for the anesthetic gases xenon, nitrous oxide, and cyclopropane. Mol. Pharmacol. 2004, 65, 443–452.
- Watts, D.; Gaete, D.; Rodriguez, D.; Hoogewijs, D.; Rauner, M.; Sormendi, S.; Wielockx, B. Hypoxia Pathway Proteins are Master Regulators of Erythropoiesis. Int. J. Mol. Sci. 2020, 21, 8131.
- Koulnis, M.; Liu, Y.; Hallstrom, K.; Socolovsky, M. Negative autoregulation by Fas stabilizes adult erythropoiesis and accelerates its stress response. PLoS ONE 2011, 6, e21192.
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 2007, 117, 1068–1077.
- Suzuki, N.; Gradin, K.; Poellinger, L.; Yamamoto, M. Regulation of hypoxia-inducible gene expression after HIF activation. Exp. Cell Res. 2017, 356, 182–186.
- Noguchi, C.T.; Asavaritikrai, P.; Teng, R.; Jia, Y. Role of erythropoietin in the brain. Crit. Rev. Oncol. Hematol. 2007, 64, 159–171.
- Dey, S.; Lee, J.; Noguchi, C.T. Erythropoietin Non-hematopoietic Tissue Response and Regulation of Metabolism during Diet Induced Obesity. Front. Pharmacol. 2021, 12, 725734.
- Egrie, J.C.; Dwyer, E.; Browne, J.K.; Hitz, A.; Lykos, M.A. Darbepoetin alfa has a longer circulating half-life and greater in vivo potency than recombinant human erythropoietin. Exp. Hematol. 2003, 31, 290–299.
- McLean, M.J.; Bukhari, A.A.; Wamil, A.W. Effects of topiramate on sodium-dependent action-potential firing by mouse spinal cord neurons in cell culture. Epilepsia 2000, 41, 21–24.
- Harding, B.; Conception, K.; Li, Y.; Zhang, L. Glucocorticoids Protect Neonatal Rat Brain in Model of Hypoxic-Ischemic Encephalopathy (HIE). Int. J. Mol. Sci. 2016, 18, 17.
- Kovacs, K.; Szakmar, E.; Meder, U.; Szakacs, L.; Cseko, A.; Vatai, B.; Szabo, A.J.; McNamara, P.J.; Szabo, M.; Jermendy, A. A Randomized Controlled Study of Low-Dose Hydrocortisone Versus Placebo in Dopamine-Treated Hypotensive Neonates Undergoing Hypothermia Treatment for Hypoxic-Ischemic Encephalopathy. J. Pediatr. 2019, 211, 13–19.e3.
- Cipolla-Neto, J.; Amaral, F.G.D. Melatonin as a Hormone: New Physiological and Clinical Insights. Endocr. Rev. 2018, 39, 990–1028.
- Chitimus, D.M.; Popescu, M.R.; Voiculescu, S.E.; Panaitescu, A.M.; Pavel, B.; Zagrean, L.; Zagrean, A.M. Melatonin’s Impact on Antioxidative and Anti-Inflammatory Reprogramming in Homeostasis and Disease. Biomolecules 2020, 10, 1211.
- Janitschke, D.; Lauer, A.A.; Bachmann, C.M.; Seyfried, M.; Grimm, H.S.; Hartmann, T.; Grimm, M.O.W. Unique Role of Caffeine Compared to Other Methylxanthines (Theobromine, Theophylline, Pentoxifylline, Propentofylline) in Regulation of AD Relevant Genes in Neuroblastoma SH-SY5Y Wild Type Cells. Int. J. Mol. Sci. 2020, 21, 9015.
- Abdel-Hady, H.; Nasef, N.; Shabaan, A.E.; Nour, I. Caffeine therapy in preterm infants. World J. Clin. Pediatr. 2015, 4, 81–93.
- Daly, J.W.; Shi, D.; Nikodijevic, O.; Jacobson, K.A. The role of adenosine receptors in the central action of caffeine. Pharmacopsychoecologia 1994, 7, 201–213.
- Echeverri, D.; Montes, F.R.; Cabrera, M.; Galan, A.; Prieto, A. Caffeine’s Vascular Mechanisms of Action. Int. J. Vasc. Med. 2010, 2010, 834060.
- Rivera-Oliver, M.; Diaz-Rios, M. Using caffeine and other adenosine receptor antagonists and agonists as therapeutic tools against neurodegenerative diseases: A review. Life Sci. 2014, 101, 1–9.
- Yang, L.; Yu, X.; Zhang, Y.; Liu, N.; Xue, X.; Fu, J. Caffeine treatment started before injury reduces hypoxic-ischemic white-matter damage in neonatal rats by regulating phenotypic microglia polarization. Pediatr. Res. 2022, 92, 1543–1554.
- Iulia, C.; Ruxandra, T.; Costin, L.B.; Liliana-Mary, V. Citicoline—A neuroprotector with proven effects on glaucomatous disease. Rom. J. Ophthalmol. 2017, 61, 152–158.
- Salamah, A.; El Amrousy, D.; Elsheikh, M.; Mehrez, M. Citicoline in hypoxic ischemic encephalopathy in neonates: A randomized controlled trial. Ital. J. Pediatr. 2023, 49, 55.
- Alvarez-Sabin, J.; Roman, G.C. The role of citicoline in neuroprotection and neurorepair in ischemic stroke. Brain Sci. 2013, 3, 1395–1414.
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593.
- Cao, G.; Gong, T.; Du, Y.; Wang, Y.; Ge, T.; Liu, J. Mechanism of metformin regulation in central nervous system: Progression and future perspectives. Biomed. Pharmacother. 2022, 156, 113686.
- Yuan, Y.; Fan, X.; Guo, Z.; Zhou, Z.; Gao, W. Metformin Protects against Spinal Cord Injury and Cell Pyroptosis via AMPK/NLRP3 Inflammasome Pathway. Anal. Cell. Pathol. 2022, 2022, 3634908.
- Ruan, C.; Guo, H.; Gao, J.; Wang, Y.; Liu, Z.; Yan, J.; Li, X.; Lv, H. Neuroprotective effects of metformin on cerebral ischemia-reperfusion injury by regulating PI3K/Akt pathway. Brain Behav. 2021, 11, e2335.
- Fang, M.; Jiang, H.; Ye, L.; Cai, C.; Hu, Y.; Pan, S.; Li, P.; Xiao, J.; Lin, Z. Metformin treatment after the hypoxia-ischemia attenuates brain injury in newborn rats. Oncotarget 2017, 8, 75308–75325.
- Sharma, S.; Nozohouri, S.; Vaidya, B.; Abbruscato, T. Repurposing metformin to treat age-related neurodegenerative disorders and ischemic stroke. Life Sci. 2021, 274, 119343.
- Lee, B.E.; Toledo, A.H.; Anaya-Prado, R.; Roach, R.R.; Toledo-Pereyra, L.H. Allopurinol, xanthine oxidase, and cardiac ischemia. J. Investig. Med. 2009, 57, 902–909.
- Kostić, D.A.; Dimitrijević, D.S.; Stojanović, G.S.; Palić, I.R.; Đorđević, A.S.; Ickovski, J.D. Xanthine Oxidase: Isolation, Assays of Activity, and Inhibition. J. Chem. 2015, 2015, 294858.
- Liu, N.; Xu, H.; Sun, Q.; Yu, X.; Chen, W.; Wei, H.; Jiang, J.; Xu, Y.; Lu, W. The Role of Oxidative Stress in Hyperuricemia and Xanthine Oxidoreductase (XOR) Inhibitors. Oxid. Med. Cell. Longev. 2021, 2021, 1470380.
- Marro, P.J.; Mishra, O.P.; Delivoria-Papadopoulos, M. Effect of allopurinol on brain adenosine levels during hypoxia in newborn piglets. Brain Res. 2006, 1073–1074, 444–450.
- Kaya, D.; Micili, S.C.; Kizmazoglu, C.; Mucuoglu, A.O.; Buyukcoban, S.; Ersoy, N.; Yilmaz, O.; Isik, A.T. Allopurinol attenuates repeated traumatic brain injury in old rats: A preliminary report. Exp. Neurol. 2022, 357, 114196.
- Goss, J.; Hair, P.; Kumar, P.; Iacono, G.; Redden, L.; Morelli, G.; Krishna, N.; Thienel, U.; Cunnion, K. RLS-0071, a dual-targeting anti-inflammatory peptide—Biomarker findings from a first in human clinical trial. Transl. Med. Commun. 2023, 8, 1.
- Soliman, H.; Theret, M.; Scott, W.; Hill, L.; Underhill, T.M.; Hinz, B.; Rossi, F.M.V. Multipotent stromal cells: One name, multiple identities. Cell Stem Cell 2021, 28, 1690–1707.
- van Velthoven, C.T.; Kavelaars, A.; Heijnen, C.J. Mesenchymal stem cells as a treatment for neonatal ischemic brain damage. Pediatr. Res. 2012, 71, 474–481.
- Ranjan, A.K.; Gulati, A. Sovateltide Mediated Endothelin B Receptors Agonism and Curbing Neurological Disorders. Int. J. Mol. Sci. 2022, 23, 3146.
- Takai, M.; Umemura, I.; Yamasaki, K.; Watakabe, T.; Fujitani, Y.; Oda, K.; Urade, Y.; Inui, T.; Yamamura, T.; Okada, T. A potent and specific agonist, Suc--endothelin-1(8-21), IRL 1620, for the ETB receptor. Biochem. Biophys. Res. Commun. 1992, 184, 953–959.
- Ramos, M.D.; Briyal, S.; Prazad, P.; Gulati, A. Neuroprotective Effect of Sovateltide (IRL 1620, PMZ 1620) in a Neonatal Rat Model of Hypoxic-Ischemic Encephalopathy. Neuroscience 2022, 480, 194–202.
- Gulati, A.; Hornick, M.G.; Briyal, S.; Lavhale, M.S. A novel neuroregenerative approach using ET(B) receptor agonist, IRL-1620, to treat CNS disorders. Physiol. Res. 2018, 67, S95–S113.
- Gulati, A.; Kumar, A.; Morrison, S.; Shahani, B.T. Effect of centrally administered endothelin agonists on systemic and regional blood circulation in the rat: Role of sympathetic nervous system. Neuropeptides 1997, 31, 301–309.
- Kaundal, R.K.; Deshpande, T.A.; Gulati, A.; Sharma, S.S. Targeting endothelin receptors for pharmacotherapy of ischemic stroke: Current scenario and future perspectives. Drug Discov. Today 2012, 17, 793–804.
- Leonard, M.G.; Briyal, S.; Gulati, A. Endothelin B receptor agonist, IRL-1620, reduces neurological damage following permanent middle cerebral artery occlusion in rats. Brain Res. 2011, 1420, 48–58.
- Leonard, M.G.; Briyal, S.; Gulati, A. Endothelin B receptor agonist, IRL-1620, provides long-term neuroprotection in cerebral ischemia in rats. Brain Res. 2012, 1464, 14–23.
- Leonard, M.G.; Prazad, P.; Puppala, B.; Gulati, A. Selective Endothelin-B Receptor Stimulation Increases Vascular Endothelial Growth Factor in the Rat Brain during Postnatal Development. Drug Res. 2015, 65, 607–613.
- Leonard, M.G.; Gulati, A. Endothelin B receptor agonist, IRL-1620, enhances angiogenesis and neurogenesis following cerebral ischemia in rats. Brain Res. 2013, 1528, 28–41.
- Ranjan, A.K.; Briyal, S.; Gulati, A. Sovateltide (IRL-1620) activates neuronal differentiation and prevents mitochondrial dysfunction in adult mammalian brains following stroke. Sci. Rep. 2020, 10, 12737.
- Ranjan, A.K.; Briyal, S.; Khandekar, D.; Gulati, A. Sovateltide (IRL-1620) affects neuronal progenitors and prevents cerebral tissue damage after ischemic stroke. Can. J. Physiol. Pharmacol. 2020, 98, 659–666.
- Briyal, S.; Ranjan, A.K.; Hornick, M.G.; Puppala, A.K.; Luu, T.; Gulati, A. Anti-apoptotic activity of ET(B) receptor agonist, IRL-1620, protects neural cells in rats with cerebral ischemia. Sci. Rep. 2019, 9, 10439.
- Cifuentes, E.G.; Hornick, M.G.; Havalad, S.; Donovan, R.L.; Gulati, A. Neuroprotective Effect of IRL-1620, an Endothelin B Receptor Agonist, on a Pediatric Rat Model of Middle Cerebral Artery Occlusion. Front. Pediatr. 2018, 6, 310.
- Gulati, A.; Agrawal, N.; Vibha, D.; Misra, U.K.; Paul, B.; Jain, D.; Pandian, J.; Borgohain, R. Safety and Efficacy of Sovateltide (IRL-1620) in a Multicenter Randomized Controlled Clinical Trial in Patients with Acute Cerebral Ischemic Stroke. CNS Drugs 2021, 35, 85–104.
- Keam, S.J. Sovateltide: First Approval. Drugs 2023, 83, 1239–1244.

