 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Artur Jorge Ferreira | -- | 2279 | 2023-10-24 09:02:31 | | | |
| 2 | Lindsay Dong | Meta information modification | 2279 | 2023-10-24 09:25:44 | | |
Video Upload Options
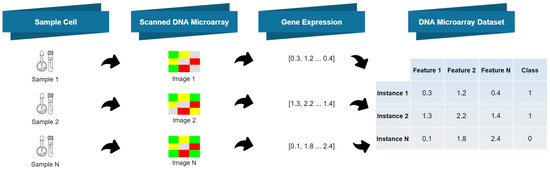
Early disease detection using microarray data is vital for prompt and efficient treatment. However, the intricate nature of these data and the ongoing need for more precise interpretation techniques make it a persistently active research field. Numerous gene expression datasets are publicly available, containing microarray data that reflect the activation status of thousands of genes in patients who may have a specific disease. ThesGene expression microarrays, also known as DNA microarrays, are laboratory tools used to measure the expression levels of thousands of genes simultaneously, thus providing a snapshot of the cellular function (for technical details.e datasets encompass a vast number of genes, resulting in high-dimensional feature vectors that present significant challenges for human analysis.
1. Introduction
2. DNA Microarrays
2.1. DNA Microarrays: Acquisition Technique and Resulting Data
-
It is composed by a solid surface, arranged in columns and rows, containing thousands of spots;
-
Each spot refers to one single gene and contains multiple strands of the same DNA, yielding a unique DNA sequence;
-
Each spot location and its corresponding DNA sequence is recorded in a database.

- 1.
-
Extraction of ribonucleic acid (RNA) from the sample cells and drawing out the messenger RNA (mRNA) from the existing RNA, because only the mRNA develops gene expression.
- 2.
-
CDNA creation: a DNA copy is made from the mRNA using the reverse transcriptase enzyme, which generates the complementary DNA (CDNA). A label is added in the CDNA representing each cell sample (e.g., with fluorescent red and green for cancer and healthy cells, respectively). This step is necessary since DNA is more stable than RNA and this labeling allows identifying the genes.
- 3.
-
Hybridization: both CDNA types are added to the DNA microarray and each spot already has many unique CDNA. When mixed together, they will base-pair each other due to the DNA complementary base pairing property. Not all CDNA strands will bind to each other, since some may not hybridize being washed off.
- 4.
-
Analysis: the DNA microarray is analyzed with a scanner to find patterns of hybridization by detecting the fluorescent colors.
-
A few red CDNA molecules bound to a spot, if the gene is expressed only in the cancer (red) cells;
-
A few green CDNA molecules bound to another spot, if the gene is expressed only in the healthy (green) cells;
-
Some of both red and green CDNA molecules bound to a single spot on the microarray, yielding a yellow spot; in this case, the gene is expressed both in the cancer and healthy cells;
-
Finally, several spots of the microarray do not have a single red or green CDNA strand bound to them; this happens if the gene is not being expressed in either type of cell.
2.2. Feature Discretization
2.3. Feature Selection
with denoting the mean of the i-th feature. In supervised mode, RRFS uses as relevance measure the Fisher ratio [14], also known as Fisher score, defined as (for the i-th feature)
where , , and are the sample means and variances of feature , for the patterns of each of the two classes (denoted as −1 and 1). This ratio measures how well each feature alone separates the two classes [14], and has been found to serve well as a relevance criterion for FS tasks. For more than two classes, FiR for feature is generalized [19][20] as
where c is the number of classes, is the number of samples in class j, and denotes the sample mean of considering only samples in class j; finally, . Among many other applications, the Fisher ratio has been used successfully with microarray data, as reported by Furey et al. [21]. When using the Fisher ratio for FS, we simply keep the top-rank features.
2.4. Classifiers
2.4.1. SVM
2.4.2. DT
2.5. Related Approaches
References
- Alonso-Betanzos, A.; Bolón-Canedo, V.; Morán-Fernández, L.; Sánchez-Marono, N. A Review of Microarray Datasets: Where to Find Them and Specific Characteristics. Methods Mol. Biol. 2019, 1986, 65–85.
- Nogueira, A.; Ferreira, A.; Figueiredo, M. A Step Towards the Explainability of Microarray Data for Cancer Diagnosis with Machine Learning Techniques. In Proceedings of the International Conference on Pattern Recognition Applications and Methods (ICPRAM), Online, 3–5 February 2022; pp. 362–369.
- Simon, R.; Korn, E.; McShane, L.; Radmacher, M.; Wright, G.; Zhao, Y. Design and Analysis of DNA Microarray Investigations; Springer: New York, NY, USA, 2003.
- Ferreira, A.; Figueiredo, M. Exploiting the bin-class histograms for feature selection on discrete data. In Proceedings of the Iberian Conference on Pattern Recognition and Image Analysis, Santiago de Compostela, Spain, 17–19 June 2015; Springer: Cham, Switzerland, 2015; pp. 345–353.
- Belkin, M.; Niyogi, P. Laplacian eigenmaps for dimensionality reduction and data representation. Neural Comput. 2003, 15, 1373–1396.
- Dougherty, J.; Kohavi, R.; Sahami, M. Supervised and unsupervised discretization of continuous features. In Machine Learning Proceedings 1995; Elsevier: Amsterdam, The Netherlands, 1995; pp. 194–202.
- Fayyad, U.; Irani, K. Multi-interval discretization of continuous-valued attributes for classification learning. In Proceedings of the International Joint Conference on Uncertainty in AI, Washington, DC, USA, 9–11 July 1993; pp. 1022–1027.
- Garcia, S.; Luengo, J.; Saez, J.; Lopez, V.; Herrera, F. A survey of discretization techniques: Taxonomy and empirical analysis in supervised learning. IEEE Trans. Knowl. Data Eng. 2013, 25, 734–750.
- Guyon, I.; Gunn, S.; Nikravesh, M.; Zadeh, L. Feature Extraction: Foundations and Applications; Springer: Berlin/Heidelberg, Germany, 2006.
- Alpaydin, E. Introduction to Machine Learning, 3rd ed.; The MIT Press: Cambridge, MA, USA, 2014.
- He, X.; Cai, D.; Niyogi, P. Laplacian score for feature selection. In Proceedings of the Advances in Neural Information Processing Systems, Vancouver, BC, Canada, 5–8 December 2005; MIT Press: Cambridge, MA, USA; Volume 18, pp. 507–514.
- Zhao, Z.; Liu, H. Spectral feature selection for supervised and unsupervised learning. In Proceedings of the 24th International Conference on Machine Learning, Corvallis, OR, USA, 20–24 June 2007; pp. 1151–1157.
- Liu, L.; Kang, J.; Yu, J.; Wang, Z. A comparative study on unsupervised feature selection methods for text clustering. In Proceedings of the 2005 International Conference on Natural Language Processing and Knowledge Engineering, Wuhan, China, 30 October–1 November 2005; IEEE: Piscataway, NJ, USA, 2005; pp. 597–601.
- Fisher, R. The use of multiple measurements in taxonomic problems. Ann. Eugen. 1936, 7, 179–188.
- Yu, L.; Liu, H. Feature selection for high-dimensional data: A fast correlation-based filter solution. In Proceedings of the International Conference on Machine Learning (ICML), Washington, DC, USA, 21–24 August 2003; pp. 856–863.
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. (PAMI) 2005, 27, 1226–1238.
- Kononenko, I. Estimating attributes: Analysis and extensions of RELIEF. In Proceedings of the European Conference on Machine Learning, Catania, Italy, 6–8 April 1994; Springer: Berlin/Heidelberg, Germany, 1994; pp. 171–182.
- Ferreira, A.; Figueiredo, M. Efficient feature selection filters for high-dimensional data. Pattern Recognit. Lett. 2012, 33, 1794–1804.
- Duda, R.; Hart, P.; Stork, D. Pattern Classification, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2001.
- Zhao, Z.; Morstatter, F.; Sharma, S.; Alelyani, S.; Anand, A.; Liu, H. Advancing Feature Selection Research—ASU Feature Selection Repository; Technical Report; Computer Science & Engineering, Arizona State University: Tempe, AZ, USA, 2010.
- Furey, T.; Cristianini, N.; Duffy, N.; Bednarski, D.; Schummer, M.; Haussler, D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics 2000, 16, 906–914.
- Alpaydin, E. Introduction to Machine Learning, 2nd ed.; The MIT Press: Cambridge, MA, USA, 2010.
- Boser, B.; Guyon, I.; Vapnik, V. A training algorithm for optimal margin classifiers. In Proceedings of the Annual ACM Workshop on Computational Learning Theory, Pittsburgh, PA, USA, 27–29 July 1992; ACM Press: New York, NY, USA, 1992; pp. 144–152.
- Burges, C. A tutorial on support vector machines for pattern recognition. Data Min. Knowl. Discov. 1998, 2, 121–167.
- Vapnik, V. The Nature of Statistical Learning Theory; Springe: New York, NY, USA, 1999.
- Hsu, C.; Lin, C. A comparison of methods for multi-class support vector machines. IEEE Trans. Neural Netw. 2002, 13, 415–425.
- Weston, J.; Watkins, C. Multi-Class Support Vector Machines; Technical Report; Department of Computer Science, Royal Holloway, University of London: London, UK, 1998.
- Breiman, L. Classification and Regression Trees, 1st ed.; Chapman & Hall/CRC: Boca Raton, FL, USA, 1984.
- Quinlan, J. Induction of decision trees. Mach. Learn. 1986, 1, 81–106.
- Quinlan, J. C4.5: Programs for Machine Learning; Morgan Kaufmann: San Mateo, CA, USA, 1993.
- Quinlan, J. Bagging, boosting, and C4.5. In Proceedings of the National Conference on Artificial Intelligence, Portland, OR, USA, 4–8 August 1996; AAAI Press: Washington, DA, USA, 1996; pp. 725–730.
- Rokach, L.; Maimon, O. Top-down induction of decision trees classifiers—A survey. IEEE Trans. Syst. Man, Cybern. Part C Appl. Rev. 2005, 35, 476–487.
- Yip, W.; Amin, S.; Li, C. A Survey of Classification Techniques for Microarray Data Analysis. In Handbook of Statistical Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2011; pp. 193–223.
- Statnikov, A.; Tsamardinos, I.; Dosbayev, Y.; Aliferis, C. GEMS: A system for automated cancer diagnosis and biomarker discovery from microarray gene expression data. Int. J. Med. Inform. 2005, 74, 491–503.
- Witten, I.; Frank, E.; Hall, M.; Pal, C. Data Mining: Practical Machine Learning Tools and Techniques, 4th ed.; Morgan Kauffmann: Mateo, CA, USA, 2016.
- Meyer, P.; Schretter, C.; Bontempi, G. Information-theoretic feature selection in microarray data using variable complementarity. IEEE J. Sel. Top. Signal Process. 2008, 2, 261–274.
- Statnikov, A.; Aliferis, C.; Tsamardinos, I.; Hardin, D.; Levy, S. A comprehensive evaluation of multicategory classification methods for microarray gene expression cancer diagnosis. Bioinformatics 2005, 21, 631–643.
- Diaz-Uriarte, R.; Andres, S. Gene selection and classification of microarray data using random forest. BMC Bioinform. 2006, 7, 3.
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32.
- Li, Z.; Xie, W.; Liu, T. Efficient feature selection and classification for microarray data. PLoS ONE 2018, 13, 0202167.
- Consiglio, A.; Casalino, G.; Castellano, G.; Grillo, G.; Perlino, E.; Vessio, G.; Licciulli, F. Explaining Ovarian Cancer Gene Expression Profiles with Fuzzy Rules and Genetic Algorithms. Electronics 2021, 10, 375.
- Saeys, Y.; Inza, I.; naga, P.L. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517.
- AbdElNabi, M.L.R.; Wajeeh Jasim, M.; El-Bakry, H.M.; Hamed, N.; Taha, M.; Khalifa, N.E.M. Breast and Colon Cancer Classification from Gene Expression Profiles Using Data Mining Techniques. Symmetry 2020, 12, 408.
- Alonso-González, C.J.; Moro-Sancho, Q.I.; Simon-Hurtado, A.; Varela-Arrabal, R. Microarray gene expression classification with few genes: Criteria to combine attribute selection and classification methods. Expert Syst. Appl. 2012, 39, 7270–7280.
- Jirapech-Umpai, T.; Aitken, S. Feature selection and classification for microarray data analysis: Evolutionary methods for identifying predictive genes. BMC Bioinform. 2005, 6, 148.

