Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Soji Morishita | -- | 2114 | 2023-10-24 08:18:11 | | | |
| 2 | Lindsay Dong | Meta information modification | 2114 | 2023-10-24 10:57:33 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Morishita, S.; Komatsu, N. Gene Alterations in BCR::ABL1-Negative Myeloproliferative Neoplasms. Encyclopedia. Available online: https://encyclopedia.pub/entry/50710 (accessed on 09 August 2026).
Morishita S, Komatsu N. Gene Alterations in BCR::ABL1-Negative Myeloproliferative Neoplasms. Encyclopedia. Available at: https://encyclopedia.pub/entry/50710. Accessed August 09, 2026.
Morishita, Soji, Norio Komatsu. "Gene Alterations in BCR::ABL1-Negative Myeloproliferative Neoplasms" Encyclopedia, https://encyclopedia.pub/entry/50710 (accessed August 09, 2026).
Morishita, S., & Komatsu, N. (2023, October 24). Gene Alterations in BCR::ABL1-Negative Myeloproliferative Neoplasms. In Encyclopedia. https://encyclopedia.pub/entry/50710
Morishita, Soji and Norio Komatsu. "Gene Alterations in BCR::ABL1-Negative Myeloproliferative Neoplasms." Encyclopedia. Web. 24 October, 2023.
Copy Citation
BCR::ABL1-negative myeloproliferative neoplasms (MPNs) are a group of hematopoietic malignancies in which somatic mutations are acquired in hematopoietic stem/progenitor cells, resulting in an abnormal increase in blood cells in peripheral blood and fibrosis in bone marrow. Mutations in JAK2, MPL, and CALR are frequently found in BCR::ABL1-negative MPNs, and detecting typical mutations in these three genes has become essential for the diagnosis of BCR::ABL1-negative MPNs. Furthermore, comprehensive gene mutation and expression analyses performed using massively parallel sequencing have identified gene mutations associated with the prognosis of BCR::ABL1-negative MPNs such as ASXL1, EZH2, IDH1/2, SRSF2, and U2AF1.
BCR::ABL1-negative myeloproliferative neoplasms
gene mutations
diagnostic marker
prognosis
1. Introduction
Myeloproliferative neoplasms (MPNs) are characterized as a clonal proliferation of hematopoietic stem/progenitor cells, which cause an increase in one or more mature myeloid lineage cells. MPNs consist of multiple subgroups: chronic myeloid leukemia (CML); polycythemia vera (PV); essential thrombocythemia (ET); prefibrotic primary myelofibrosis (PMF); overt PMF; chronic neutrophilic leukemia (CNL); chronic eosinophilic leukemia (CEL); and unclassifiable MPN, not otherwise specified (Figure 1) [1][2]. In general, CML involves a typical driver gene alteration, the BCR::ABL1 fusion gene, and more rare diseases, namely CNL, CEL, and unclassifiable MPN, not otherwise specified, have been treated as independent diseases. In contrast, patients with BCR::ABL1-negative MPNs (PV, ET, prefibrotic PMF, and overt PMF) transform to other subgroups with a 10–15% frequency and share common gene mutations, namely JAK2 mutations (V617F and exon 12), MPLW515L/K, and CALR exon 9 frameshift mutations, in a mutually exclusive manner [3][4][5][6][7][8][9].
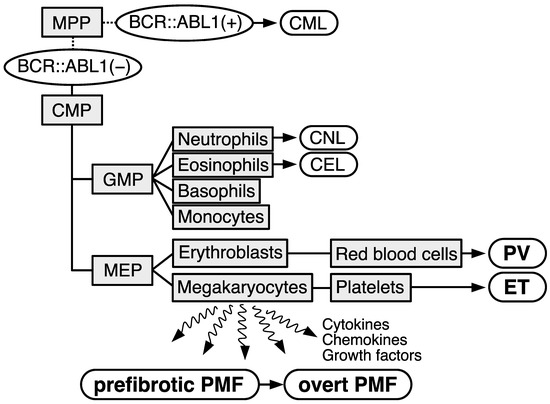
Figure 1. Schematical illustration describing the subtypes of myeloproliferative neoplasms (MPNs). Cell types are depicted as light gray squares. MPP: multipotent progenitor; CMP: common myeloid progenitor; GMP: granulocyte-monocyte progenitor; MEP: megakaryocyte–erythrocyte progenitor. Subtypes of MPNs are depicted as white rounded rectangles. Chronic myeloid leukemia (CML) exhibits BCR::ABL1 gene (BCR::ABL1(+)). Other subtypes are stratified as BCR::ABL1-negative (−) MPNs. CNL: chronic neutrophilic leukemia; CEL: chronic eosinophilic leukemia; PV: polycythemia vera; ET: essential thrombocythemia; PMF: primary myelofibrosis.
2. JAK2 Mutations
The JAK2 mutations considered as driver mutations of BCR::ABL1-negative MPNs are V617F substitution and complex mutations, including missense and in-frame deletions/insertions at exon 12 [3][4][5][6]. These mutations concentrate around the JH2 domain, which suppresses the kinase activity of the JH1 domain in JAK2 under a static state. JAK2 mutations decrease the suppression of kinase activity by the JH2 domain, resulting in the constitutive activation of JAK2. The JAK2V617F mutation is a single nucleotide alteration from guanine to thymine at nucleotide position 1849, which causes an amino acid change from V (valine, GTC) to F (phenylalanine, TTC) at codon 617. In addition, the JAK2V617F mutation was initially identified from the three driver gene mutations of BCR::ABL1-negative MPNs [3][4][5] and has been most frequently identified among the patients with BCR::ABL1-negative MPNs, and the positivity is approximately 97% in PV and approximately 50% in ET and PMF. In contrast, JAK2 exon 12 mutations are specific for PV, with 3% positivity, and a variety of mutations have been identified at JAK2 exon 12 (Supplementary Table S1, according to the Catalogue Of Somatic Mutations In Cancer (COSMIC) database, as of June 2023) [10][11]. Clinically, patients with PV harboring the JAK2V617F mutation exhibit pancytosis, including leukocytosis, thrombocytosis, and erythrocytosis, whereas those harboring the JAK2 exon 12 mutation show only an aggressive increase in red cell mass. As for the relationship between the prognosis and JAK2 mutations, no significant differences between JAK2V617F-mutated and JAK2 exon 12-mutated patients with PV have been observed [12]; nevertheless, JAK2V617F is a well-known risk factor of thrombosis [13][14]. Thrombosis promoted through the increased neutrophil extracellular trap formation was observed in the JAK2V617F-mutated murine model [15]. JAK2V617F is also useful for monitoring the efficacy of treatments or predicting the outcome of patients. Pegylated interferon-α, for example, is one of the recently developed drugs against MPNs and decreases the JAK2V617F allele burden in patients [16][17][18].
3. MPL Mutations
The majority of MPL mutations involve the substitution of W (tryptophane, c.1542-1544TGG) at codon 515 with other nucleic acids, causing W515L/K/A/R (leucine/lysine/alanine/arginine) mutations [7][19]. In addition to these W515 mutations, the substitution of S (serine) at codon 505 with N/C (asparagine/cysteine) has been identified [20][21]. These mutations are located at the membrane-spanning segment of MPL and are considered to involve conformation changes that trigger constitutive activation of the downstream molecules. Although the frequency of MPL mutations in BCR::ABL1-negative MPNs is low (5% at most in ET, <10% in PMF), considering that mutant CALR binds to MPL and activates downstream signals [22], signal activation through MPL may play a key role in the pathogenesis of BCR::ABL1-negative MPNs [23].
4. CALR Exon 9 Frameshift Mutations
The CALR mutation is the most recently discovered among the driver gene mutations in BCR::ABL1-negative MPNs and is found in approximately 20–30% of patients with ET and PMF [8][9]. This mutation is characterized by the presence of a deletion or insertion at the end of exon 9, the final exon of the CALR gene. Over 100 variations have been found according to the COSMIC database, as of January 2023), all of which cause the same frameshift and produce a common amino acid sequence at the C-terminus when translated into protein. Among them, deletions of 52 bases (type 1, p.L367fs*46) and insertions of 5 bases (type 2, p.K385fs*47) are the major mutations, accounting for approximately 85% of the observed variations. Mutant CALR activates downstream signaling by forming homomultimeric complexes through a new amino acid sequence generated by the mutation, changing the structure of the CALR protein and allowing it to bind with MPL [22][24][25]. Patients with overt PMF harboring a CALR mutation have a better prognosis than those with other driver gene mutations [26]. Regarding the CALR mutations in overt PMF, CALR type 1 mutations are dominant, and patients harboring the type 2 mutation exhibit a poorer prognosis than those with the type 1 mutation [27][28].
5. Triple-Negative BCR::ABL1-Negative MPNs
A portion of patients with BCR::ABL1-negative MPNs (none or rare in PV, 10–15% in ET, and ~10% in PMF) have none of the driver mutations, referred to as TN cases. Noncanonical somatic mutations at driver genes of BCR::ABL1-negative MPNs (e.g., JAK2G571S and MPLS204F/P) have been identified in TN cases; however, it should be considered that these mutations do not account for all the remaining cases [29][30] and no evidence of cytokine-independent cell growth has been reported for these mutations. Whole exome sequencing and analysis of TN-ET have shown that approximately half of the patients exhibited polyclonal cell differentiation, which implies that some cases of thrombocytosis in TN-ET may be caused by nonneoplastic diseases [29][31].
6. CREB3L1 as a Novel Diagnostic Marker of BCR::ABL1-Negative MPNs
To diagnose TN cases in practice, a histopathological diagnosis of the BM biopsy is required. However, the discrimination of TN from reactive cases is challenging because the pathological diagnosis of BCR::ABL1-negative MPNs is not always reproducible, even for expert hematopathologists. By focusing on the fact that typical clinical presentations (i.e., thrombocytosis) of ET are similar regardless of the presence and type of driver gene mutations, and that the downstream RNA expression may be common and different from that in reactive cases, differential expression analysis utilizing RNA from platelet-rich plasma (PRP) obtained from ET and reactive thrombocytosis patients was conducted. As a result, CREB3L1 was found to be specifically overexpressed among ET patients [32]. CREB3L1 is a transcription factor that localizes in the endoplasmic reticulum (ER), migrates into the nucleus in response to ER stress, and induces the expression of various genes [33]. Although the role of CREB3L1 in the pathogenesis of BCR::ABL1-negative MPNs remains unclear, the IRE1a/XBP1 pathway, which is an ER stress-responsible pathway other than CREB3L1, is activated by the CALR type 1 mutation and drives BCR::ABL1-negative MPNs [34].
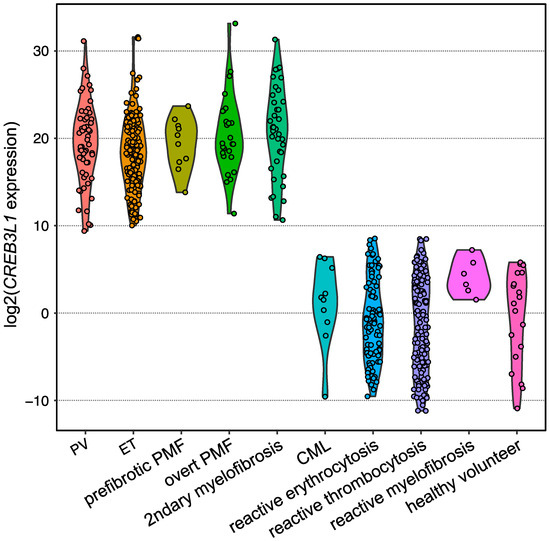
Expansion of the testing of CREB3L1 overexpression for other subtypes of BCR::ABL1-negative MPNs harboring one of the driver gene mutations in the validation analysis by employing quantitative PCR revealed that CREB3L1 was overexpressed widely among BCR::ABL1-negative MPNs compared with those of reactive cases and healthy volunteers (Figure 2). The area under the ROC curve showed that the sensitivity and specificity were both 1.0000, indicating that the CREB3L1 overexpression in PRP discriminates driver gene-mutated BCR::ABL1-negative MPNs from reactive cases [32]. Further investigations are required to determine whether CREB3L1 expression is a diagnostic marker for BCR::ABL1-negative MPNs, including TN cases.
Figure 2. Violin plot showing the expression levels of CREB3L1 measured using reverse transcription quantitative PCR. Dots represent the CREB3L1 levels of the individuals, which are expressed as the value relative to the mean expression levels among healthy volunteers. B2M was used as an internal control.
7. Nondriver Mutations and Their Association with the Prognosis of BCR::ABL1-Negative MPNs
Comprehensive genome analyses, such as MPS, have identified some mutations relating to epigenetic modification and RNA-splicing among patients with BCR::ABL1-negative MPNs at low frequency [35][36]. MPS-based comprehensive target resequencing methodology focusing on these nondriver mutations also revealed that the mutations highly accumulated in patients with BCR::ABL1-negative MPNs were correlated with a poor prognosis. Based on these findings, mutation-enhanced prognostic scoring systems based on the positivity of nondriver mutations have been proposed and are widely used to estimate the prognostic risks of patients with BCR::ABL1-negative MPNs.
Similar to the results obtained by other groups, patients with PMF harboring ASXL1, EZH2, and/or SRSF2 mutations exhibited significantly shorter 5-year overall survival, and these gene mutations are also the poor prognostic factors of PMF that were demonstrated in the Japanese cohort [37]. Furthermore, regarding ET and PV, the frequencies of ASXL1 and EZH2 mutations increase as the diseases progresses from ET or PV to prefibrotic PMF and overt PMF, whereas the frequencies of DNMT3A and TET2 mutations are unrelated to disease type. This implies that ASXL1 and EZH2 mutations are related to disease progression, whereas DNMT3A and TET2 mutations may trigger the disease. Logistic regression analysis showed that ASXL1 mutation-positive ET/PV patients had a high rate of progression to leukemia and myelofibrosis [38].
8. Genetic Background Enhancing the Risk of Developing BCR::ABL1-Negative MPNs
The accumulation of mutations is highly dependent on age, and elderly individuals sometimes develop clonal hematopoiesis because of acquired mutations [39][40]. Such individuals are diagnosed with clonal hematopoiesis of intermediate potential (CHIP) or age-related clonal hematopoiesis (ARCH) [41][42]. Regarding the development of CHIP/ARCH, it has been experimentally demonstrated using the zebrafish model that mutant clones increase by developing clonal fitness, which is driven by enhanced resistance to inflammatory signals [43]. Notably, JAK2V617F has been identified with a frequency of approximately 0.1% in an analysis of 49,488 individuals, and 7 patients harboring JAK2V617F (48 individuals were removed for originally having MPNs) were then considered as CHIP/ARCH in the final cohort. Furthermore, the JAK2V617F mutant burden in CHIP/ARCH increases by 0.55% per year in the study, implying that the JAK2V617F-mutated cells acquire a mild growth advantage, and therefore the development of BCR::ABL1-negative MPNs may progress over time [44]. This implication is supported by another investigation, which clarified that JAK2V617F mutations occur decades before BCR::ABL1-negative MPN diagnosis, increase the fitness of HSCs, and induce a megakaryocyte–erythroid differentiation bias [45]. CHIP/ARCH individuals also have an increased risk of developing hematologic malignancies or cardiovascular diseases in patients with additional mutations [46].
Some studies have investigated the impact of genetic background on the risk of developing BCR::ABL1-negative MPNs. For example, the JAK2 46/1 haplotype concomitant with JAK2V617F, RBBP6, SH2B3, and TERT mutations was shown to increase the risk of developing BCR::ABL1-negative MPNs [47][48][49][50][51][52]. A GWAS analysis of 888,503 individuals, including 2949 patients with BCR::ABL1-negative MPNs, identified 17 loci, including JAK2 and TERT [53]. SNPs located at the identified loci, such as rs17879961 (located at CHEK2 exon 5) and rs534137 (located at the promoter region of GFI1B), were considered to induce the instability of HSCs homeostasis and may predispose patients to BCR::ABL1-negative MPNs.
In addition to SNPs that confer higher susceptibility to develop BCR::ABL1-negative MPNs, SNPs that affect the phenotype, prognosis, and response to therapies of BCR::ABL1-negative MPNs have been reported and summarized [54]. Although allelic frequencies are not rare (0.114822 for rs6198, 0.279039 for rs1024611, and 0.444187 for rs2431697 by gnomAD, respectively), a poorer prognosis was observed among patients with PMF harboring both JAK2V617F and homozygous mutations of rs6198 locating at NR3C1 than those bearing wild-type NR3C1 [55], rs1024611 at CCL2 strongly correlated to the CCL2 expression and the myelofibrosis grade [56], and homozygous rs2431697 at miR-146a was associated with myelofibrosis progression [57].
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228.
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719.
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790.
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061.
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148.
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468.
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476.
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405.
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390.
- Percy, M.J.; Scott, L.M.; Erber, W.N.; Harrison, C.N.; Reilly, J.T.; Jones, F.G.; Green, A.R.; McMullin, M.F. The frequency of JAK2 exon 12 mutations in idiopathic erythrocytosis patients with low serum erythropoietin levels. Haematologica 2007, 92, 1607–1614.
- Scott, L.M.; Beer, P.A.; Bench, A.J.; Erber, W.N.; Green, A.R. Prevalance of JAK2 V617F and exon 12 mutations in polycythaemia vera. Br. J. Haematol. 2007, 139, 511–512.
- Tondeur, S.; Paul, F.; Riou, J.; Mansier, O.; Ranta, D.; Le Clech, L.; Lippert, E.; Tavitian, S.; Chaoui, D.; Mercier, M.; et al. Long-term follow-up of JAK2 exon 12 polycythemia vera: A French Intergroup of Myeloproliferative Neoplasms (FIM) study. Leukemia 2020, 35, 871–875.
- Barbui, T.; Finazzi, G.; Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood 2012, 120, 5128–5133.
- Barbui, T.; Vannucchi, A.M.; Buxhofer-Ausch, V.; De Stefano, V.; Betti, S.; Rambaldi, A.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J. 2015, 5, e369.
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, eaan8292.
- Verstovsek, S.; Komatsu, N.; Gill, H.; Jin, J.; Lee, S.E.; Hou, H.A.; Sato, T.; Qin, A.; Urbanski, R.; Shih, W.; et al. SURPASS-ET: Phase III study of ropeginterferon alfa-2b versus anagrelide as second-line therapy in essential thrombocythemia. Future Oncol. 2022, 18, 2999–3009.
- Mesa, R.; Komatsu, N.; Gill, H.; Jin, J.; Lee, S.E.; Hou, H.A.; Sato, T.; Qin, A.; Urbanski, R.; Shih, W.; et al. MPN-545 Surpass-ET: Ropeginterferon Alfa-2b (P1101) vs. Anagrelide as Second Line Therapy in Essential Thrombocythemia. Clin. Lymphoma Myeloma Leuk. 2022, 22 (Suppl. S2), S342.
- Kiladjian, J.J.; Klade, C.; Georgiev, P.; Krochmalczyk, D.; Gercheva-Kyuchukova, L.; Egyed, M.; Dulicek, P.; Illes, A.; Pylypenko, H.; Sivcheva, L.; et al. Long-term outcomes of polycythemia vera patients treated with ropeginterferon Alfa-2b. Leukemia 2022, 36, 1408–1411.
- Boyd, E.M.; Bench, A.J.; Goday-Fernandez, A.; Anand, S.; Vaghela, K.J.; Beer, P.; Scott, M.A.; Bareford, D.; Green, A.R.; Huntly, B.; et al. Clinical utility of routine MPL exon 10 analysis in the diagnosis of essential thrombocythaemia and primary myelofibrosis. Br. J. Haematol. 2010, 149, 250–257.
- Pietra, D.; Brisci, A.; Rumi, E.; Boggi, S.; Elena, C.; Pietrelli, A.; Bordoni, R.; Ferrari, M.; Passamonti, F.; De Bellis, G.; et al. Deep sequencing reveals double mutations in cis of MPL exon 10 in myeloproliferative neoplasms. Haematologica 2011, 96, 607–611.
- Beer, P.A.; Campbell, P.J.; Scott, L.M.; Bench, A.J.; Erber, W.N.; Bareford, D.; Wilkins, B.S.; Reilly, J.T.; Hasselbalch, H.C.; Bowman, R.; et al. MPL mutations in myeloproliferative disorders: Analysis of the PT-1 cohort. Blood 2008, 112, 141–149.
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 2016, 127, 1307–1316.
- Cui, L.; Moraga, I.; Lerbs, T.; Van Neste, C.; Wilmes, S.; Tsutsumi, N.; Trotman-Grant, A.C.; Gakovic, M.; Andrews, S.; Gotlib, J.; et al. Tuning MPL signaling to influence hematopoietic stem cell differentiation and inhibit essential thrombocythemia progenitors. Proc. Natl. Acad. Sci. USA 2021, 118, e2017849118.
- Araki, M.; Yang, Y.; Imai, M.; Mizukami, Y.; Kihara, Y.; Sunami, Y.; Masubuchi, N.; Edahiro, Y.; Hironaka, Y.; Osaga, S.; et al. Homomultimerization of mutant calreticulin is a prerequisite for MPL binding and activation. Leukemia 2019, 33, 122–131.
- Masubuchi, N.; Araki, M.; Yang, Y.; Hayashi, E.; Imai, M.; Edahiro, Y.; Hironaka, Y.; Mizukami, Y.; Kihara, Y.; Takei, H.; et al. Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia 2020, 34, 499–509.
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martinez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069.
- Tefferi, A.; Lasho, T.L.; Tischer, A.; Wassie, E.A.; Finke, C.M.; Belachew, A.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.D. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood 2014, 124, 2465–2466.
- Tefferi, A.; Lasho, T.L.; Finke, C.; Belachew, A.A.; Wassie, E.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A. Type 1 vs type 2 calreticulin mutations in primary myelofibrosis: Differences in phenotype and prognostic impact. Leukemia 2014, 28, 1568–1570.
- Milosevic Feenstra, J.D.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332.
- Ma, W.; Kantarjian, H.; Zhang, X.; Yeh, C.H.; Zhang, Z.J.; Verstovsek, S.; Albitar, M. Mutation profile of JAK2 transcripts in patients with chronic myeloproliferative neoplasias. J. Mol. Diagn. 2009, 11, 49–53.
- Cabagnols, X.; Favale, F.; Pasquier, F.; Messaoudi, K.; Defour, J.P.; Ianotto, J.C.; Marzac, C.; Le Couedic, J.P.; Droin, N.; Chachoua, I.; et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood 2016, 127, 333–342.
- Morishita, S.; Yasuda, H.; Yamawaki, S.; Kawaji, H.; Itoh, M.; Edahiro, Y.; Imai, M.; Kogo, Y.; Tsuneda, S.; Ohsaka, A.; et al. CREB3L1 overexpression as a potential diagnostic marker of Philadelphia chromosome-negative myeloproliferative neoplasms. Cancer Sci. 2021, 112, 884–892.
- Sampieri, L.; Di Giusto, P.; Alvarez, C. CREB3 Transcription Factors: ER-Golgi Stress Transducers as Hubs for Cellular Homeostasis. Front. Cell Dev. Biol. 2019, 7, 123.
- Ibarra, J.; Elbanna, Y.A.; Kurylowicz, K.; Ciboddo, M.; Greenbaum, H.S.; Arellano, N.S.; Rodriguez, D.; Evers, M.; Bock-Hughes, A.; Liu, C.; et al. Type I but Not Type II Calreticulin Mutations Activate the IRE1alpha/XBP1 Pathway of the Unfolded Protein Response to Drive Myeloproliferative Neoplasms. Blood Cancer Discov. 2022, 3, 298–315.
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111.
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30.
- Morishita, S.; Ochiai, T.; Misawa, K.; Osaga, S.; Inano, T.; Fukuda, Y.; Edahiro, Y.; Ohsaka, A.; Araki, M.; Komatsu, N. Clinical impacts of the mutational spectrum in Japanese patients with primary myelofibrosis. Int. J. Hematol. 2021, 113, 500–507.
- Morishita, S.; Hashimoto, Y.; Furuya, C.; Edahiro, Y.; Ochiai, T.; Shirane, S.; Inano, T.; Yasuda, H.; Ando, M.; Araki, M.; et al. Non-driver gene mutation analysis in a large cohort of polycythemia vera and essential thrombocythemia. Eur. J. Haematol. 2023, 110, 131–136.
- Kessler, M.D.; Damask, A.; O’Keeffe, S.; Banerjee, N.; Li, D.; Watanabe, K.; Marketta, A.; Van Meter, M.; Semrau, S.; Horowitz, J.; et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 2022, 612, 301–309.
- Kar, S.P.; Quiros, P.M.; Gu, M.; Jiang, T.; Mitchell, J.; Langdon, R.; Iyer, V.; Barcena, C.; Vijayabaskar, M.S.; Fabre, M.A.; et al. Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nat. Genet. 2022, 54, 1155–1166.
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487.
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498.
- Avagyan, S.; Henninger, J.E.; Mannherz, W.P.; Mistry, M.; Yoon, J.; Yang, S.; Weber, M.C.; Moore, J.L.; Zon, L.I. Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science 2021, 374, 768–772.
- Nielsen, C.; Bojesen, S.E.; Nordestgaard, B.G.; Kofoed, K.F.; Birgens, H.S. JAK2V617F somatic mutation in the general population: Myeloproliferative neoplasm development and progression rate. Haematologica 2014, 99, 1448–1455.
- Van Egeren, D.; Escabi, J.; Nguyen, M.; Liu, S.; Reilly, C.R.; Patel, S.; Kamaz, B.; Kalyva, M.; DeAngelo, D.J.; Galinsky, I.; et al. Reconstructing the Lineage Histories and Differentiation Trajectories of Individual Cancer Cells in Myeloproliferative Neoplasms. Cell Stem Cell 2021, 28, 514–523.e9.
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, eaan4673.
- Jones, A.V.; Chase, A.; Silver, R.T.; Oscier, D.; Zoi, K.; Wang, Y.L.; Cario, H.; Pahl, H.L.; Collins, A.; Reiter, A.; et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat. Genet. 2009, 41, 446–449.
- Jager, R.; Harutyunyan, A.S.; Rumi, E.; Pietra, D.; Berg, T.; Olcaydu, D.; Houlston, R.S.; Cazzola, M.; Kralovics, R. Common germline variation at the TERT locus contributes to familial clustering of myeloproliferative neoplasms. Am. J. Hematol. 2014, 89, 1107–1110.
- Tapper, W.; Jones, A.V.; Kralovics, R.; Harutyunyan, A.S.; Zoi, K.; Leung, W.; Godfrey, A.L.; Guglielmelli, P.; Callaway, A.; Ward, D.; et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat. Commun. 2015, 6, 6691.
- Trifa, A.P.; Banescu, C.; Tevet, M.; Bojan, A.; Dima, D.; Urian, L.; Torok-Vistai, T.; Popov, V.M.; Zdrenghea, M.; Petrov, L.; et al. TERT rs2736100 A>C SNP and JAK2 46/1 haplotype significantly contribute to the occurrence of JAK2 V617F and CALR mutated myeloproliferative neoplasms—A multicentric study on 529 patients. Br. J. Haematol. 2016, 174, 218–226.
- Harutyunyan, A.S.; Giambruno, R.; Krendl, C.; Stukalov, A.; Klampfl, T.; Berg, T.; Chen, D.; Milosevic Feenstra, J.D.; Jager, R.; Gisslinger, B.; et al. Germline RBBP6 mutations in familial myeloproliferative neoplasms. Blood 2016, 127, 362–365.
- Rumi, E.; Harutyunyan, A.S.; Pietra, D.; Feenstra, J.D.; Cavalloni, C.; Roncoroni, E.; Casetti, I.; Bellini, M.; Milanesi, C.; Renna, M.C.; et al. LNK mutations in familial myeloproliferative neoplasms. Blood 2016, 128, 144–145.
- Bao, E.L.; Nandakumar, S.K.; Liao, X.; Bick, A.G.; Karjalainen, J.; Tabaka, M.; Gan, O.I.; Havulinna, A.S.; Kiiskinen, T.T.J.; Lareau, C.A.; et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature 2020, 586, 769–775.
- Masselli, E.; Pozzi, G.; Carubbi, C.; Vitale, M. The Genetic Makeup of Myeloproliferative Neoplasms: Role of Germline Variants in Defining Disease Risk, Phenotypic Diversity and Outcome. Cells 2021, 10, 2597.
- Poletto, V.; Rosti, V.; Villani, L.; Catarsi, P.; Carolei, A.; Campanelli, R.; Massa, M.; Martinetti, M.; Viarengo, G.; Malovini, A.; et al. A3669G polymorphism of glucocorticoid receptor is a susceptibility allele for primary myelofibrosis and contributes to phenotypic diversity and blast transformation. Blood 2012, 120, 3112–3117.
- Hodeib, H.; Hai, D.A.; Tawfik, M.A.; Allam, A.A.; Selim, A.; Elsawy, A.A.; Youssef, A. CCL2 rs1024611Gene Polymorphism in Philadelphia-Negative Myeloproliferative Neoplasms. Genes 2022, 13, 492.
- Ferrer-Marin, F.; Arroyo, A.B.; Bellosillo, B.; Cuenca, E.J.; Zamora, L.; Hernandez-Rivas, J.M.; Hernandez-Boluda, J.C.; Fernandez-Rodriguez, C.; Luno, E.; Hernandez, C.G.; et al. miR-146a rs2431697 identifies myeloproliferative neoplasm patients with higher secondary myelofibrosis progression risk. Leukemia 2020, 34, 2648–2659.
More
Information
Subjects:
Hematology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
461
Revisions:
2 times
(View History)
Update Date:
24 Oct 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No