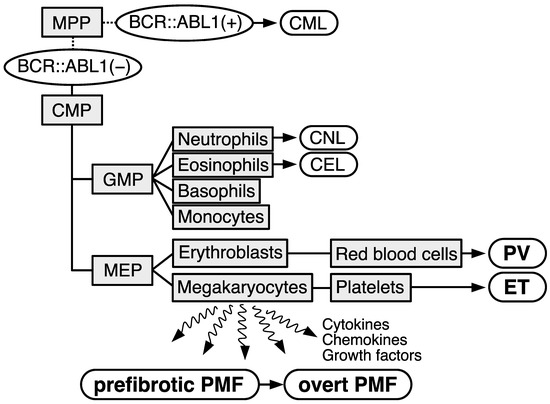
The
JAK2 mutations considered as driver mutations of
BCR::ABL1-negative MPNs are V617F substitution and complex mutations, including missense and in-frame deletions/insertions at exon 12 [
3,
4,
5,
6]. These mutations concentrate around the JH2 domain, which suppresses the kinase activity of the JH1 domain in JAK2 under a static state.
JAK2 mutations decrease the suppression of kinase activity by the JH2 domain, resulting in the constitutive activation of JAK2. The
JAK2V617F mutation is a single nucleotide alteration from guanine to thymine at nucleotide position 1849, which causes an amino acid change from V (valine, GTC) to F (phenylalanine, TTC) at codon 617. In addition, the
JAK2V617F mutation was initially identified from the three driver gene mutations of
BCR::ABL1-negative MPNs [
3,
4,
5] and has been most frequently identified among the patients with
BCR::ABL1-negative MPNs, and the positivity is approximately 97% in PV and approximately 50% in ET and PMF. In contrast,
JAK2 exon 12 mutations are specific for PV, with 3% positivity, and a variety of mutations have been identified at
JAK2 exon 12 (
Supplementary Table S1, according to the Catalogue Of Somatic Mutations In Cancer (COSMIC) database, as of June 2023) [
20,
21]. Clinically, patients with PV harboring the
JAK2V617F mutation exhibit pancytosis, including leukocytosis, thrombocytosis, and erythrocytosis, whereas those harboring the
JAK2 exon 12 mutation show only an aggressive increase in red cell mass. As for the relationship between the prognosis and
JAK2 mutations, no significant differences between
JAK2V617F-mutated and
JAK2 exon 12-mutated patients with PV have been observed [
22]; nevertheless,
JAK2V617F is a well-known risk factor of thrombosis [
23,
24]. Thrombosis promoted through the increased neutrophil extracellular trap formation was observed in the
JAK2V617F-mutated murine model [
25].
JAK2V617F is also useful for monitoring the efficacy of treatments or predicting the outcome of patients. Pegylated interferon-α, for example, is one of the recently developed drugs against MPNs and decreases the
JAK2V617F allele burden in patients [
26,
27,
28].
3. MPL Mutations
The majority of MPL mutations involve the substitution of W (tryptophane, c.1542-1544TGG) at codon 515 with other nucleic acids, causing W515L/K/A/R (leucine/lysine/alanine/arginine) mutations [
7,
34]. In addition to these W515 mutations, the substitution of S (serine) at codon 505 with N/C (asparagine/cysteine) has been identified [
35,
36]. These mutations are located at the membrane-spanning segment of MPL and are considered to involve conformation changes that trigger constitutive activation of the downstream molecules. Although the frequency of MPL mutations in
BCR::ABL1-negative MPNs is low (5% at most in ET, <10% in PMF), considering that mutant CALR binds to MPL and activates downstream signals [
37], signal activation through MPL may play a key role in the pathogenesis of
BCR::ABL1-negative MPNs [
38].
4. CALR Exon 9 Frameshift Mutations
The
CALR mutation is the most recently discovered among the driver gene mutations in
BCR::ABL1-negative MPNs and is found in approximately 20–30% of patients with ET and PMF [
8,
9]. This mutation is characterized by the presence of a deletion or insertion at the end of exon 9, the final exon of the
CALR gene. Over 100 variations have been found according to the COSMIC database, as of January 2023), all of which cause the same frameshift and produce a common amino acid sequence at the C-terminus when translated into protein. Among them, deletions of 52 bases (type 1, p.L367fs*46) and insertions of 5 bases (type 2, p.K385fs*47) are the major mutations, accounting for approximately 85% of the observed variations. Mutant CALR activates downstream signaling by forming homomultimeric complexes through a new amino acid sequence generated by the mutation, changing the structure of the CALR protein and allowing it to bind with MPL [
37,
41,
42]. Patients with overt PMF harboring a
CALR mutation have a better prognosis than those with other driver gene mutations [
43]. Regarding the
CALR mutations in overt PMF,
CALR type 1 mutations are dominant, and patients harboring the type 2 mutation exhibit a poorer prognosis than those with the type 1 mutation [
44,
45].
5. Triple-Negative BCR::ABL1-Negative MPNs
A portion of patients with
BCR::ABL1-negative MPNs (none or rare in PV, 10–15% in ET, and ~10% in PMF) have none of the driver mutations, referred to as TN cases. Noncanonical somatic mutations at driver genes of
BCR::ABL1-negative MPNs (e.g.,
JAK2G571S and
MPLS204F/P) have been identified in TN cases; however, it should be considered that these mutations do not account for all the remaining cases [
12,
46] and no evidence of cytokine-independent cell growth has been reported for these mutations. Whole exome sequencing and analysis of TN-ET have shown that approximately half of the patients exhibited polyclonal cell differentiation, which implies that some cases of thrombocytosis in TN-ET may be caused by nonneoplastic diseases [
12,
47].
6. CREB3L1 as a Novel Diagnostic Marker of BCR::ABL1-Negative MPNs
To diagnose TN cases in practice, a histopathological diagnosis of the BM biopsy is required. However, the discrimination of TN from reactive cases is challenging because the pathological diagnosis of
BCR::ABL1-negative MPNs is not always reproducible, even for expert hematopathologists. By focusing on the fact that typical clinical presentations (i.e., thrombocytosis) of ET are similar regardless of the presence and type of driver gene mutations, and that the downstream RNA expression may be common and different from that in reactive cases, differential expression analysis utilizing RNA from platelet-rich plasma (PRP) obtained from ET and reactive thrombocytosis patients was conducted. As a result,
CREB3L1 was found to be specifically overexpressed among ET patients [
13]. CREB3L1 is a transcription factor that localizes in the endoplasmic reticulum (ER), migrates into the nucleus in response to ER stress, and induces the expression of various genes [
49]. Although the role of CREB3L1 in the pathogenesis of
BCR::ABL1-negative MPNs remains unclear, the IRE1a/XBP1 pathway, which is an ER stress-responsible pathway other than CREB3L1, is activated by the CALR type 1 mutation and drives
BCR::ABL1-negative MPNs [
50].
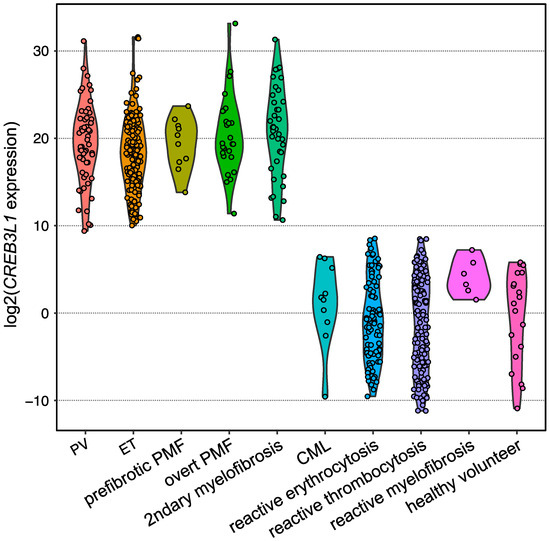
Expansion of the testing of
CREB3L1 overexpression for other subtypes of
BCR::ABL1-negative MPNs harboring one of the driver gene mutations in the validation analysis by employing quantitative PCR revealed that
CREB3L1 was overexpressed widely among
BCR::ABL1-negative MPNs compared with those of reactive cases and healthy volunteers (
Figure 5). The area under the ROC curve showed that the sensitivity and specificity were both 1.0000, indicating that the
CREB3L1 overexpression in PRP discriminates driver gene-mutated
BCR::ABL1-negative MPNs from reactive cases [
13]. Further investigations are required to determine whether
CREB3L1 expression is a diagnostic marker for
BCR::ABL1-negative MPNs, including TN cases.
Figure 5. Violin plot showing the expression levels of
CREB3L1 measured using reverse transcription quantitative PCR. Dots represent the
CREB3L1 levels of the individuals, which are expressed as the value relative to the mean expression levels among healthy volunteers.
B2M was used as an internal control.
7. Nondriver Mutations and Their Association with the Prognosis of BCR::ABL1-Negative MPNs
Comprehensive genome analyses, such as MPS, have identified some mutations relating to epigenetic modification and RNA-splicing among patients with
BCR::ABL1-negative MPNs at low frequency [
14,
15]. MPS-based comprehensive target resequencing methodology focusing on these nondriver mutations also revealed that the mutations highly accumulated in patients with
BCR::ABL1-negative MPNs were correlated with a poor prognosis. Based on these findings, mutation-enhanced prognostic scoring systems based on the positivity of nondriver mutations have been proposed and are widely used to estimate the prognostic risks of patients with
BCR::ABL1-negative MPNs.
Similar to the results obtained by other groups, patients with PMF harboring
ASXL1,
EZH2, and/or
SRSF2 mutations exhibited significantly shorter 5-year overall survival, and these gene mutations are also the poor prognostic factors of PMF that were demonstrated in the Japanese cohort [
16]. Furthermore, regarding ET and PV, the frequencies of
ASXL1 and
EZH2 mutations increase as the diseases progresses from ET or PV to prefibrotic PMF and overt PMF, whereas the frequencies of
DNMT3A and
TET2 mutations are unrelated to disease type. This implies that
ASXL1 and
EZH2 mutations are related to disease progression, whereas
DNMT3A and
TET2 mutations may trigger the disease. Logistic regression analysis showed that
ASXL1 mutation-positive ET/PV patients had a high rate of progression to leukemia and myelofibrosis [
17].
8. Genetic Background Enhancing the Risk of Developing BCR::ABL1-Negative MPNs
The accumulation of mutations is highly dependent on age, and elderly individuals sometimes develop clonal hematopoiesis because of acquired mutations [
61,
62]. Such individuals are diagnosed with clonal hematopoiesis of intermediate potential (CHIP) or age-related clonal hematopoiesis (ARCH) [
63,
64]. Regarding the development of CHIP/ARCH, it has been experimentally demonstrated using the zebrafish model that mutant clones increase by developing clonal fitness, which is driven by enhanced resistance to inflammatory signals [
65]. Notably,
JAK2V617F has been identified with a frequency of approximately 0.1% in an analysis of 49,488 individuals, and 7 patients harboring
JAK2V617F (48 individuals were removed for originally having MPNs) were then considered as CHIP/ARCH in the final cohort. Furthermore, the
JAK2V617F mutant burden in CHIP/ARCH increases by 0.55% per year in the study, implying that the
JAK2V617F-mutated cells acquire a mild growth advantage, and therefore the development of
BCR::ABL1-negative MPNs may progress over time [
66]. This implication is supported by another investigation, which clarified that
JAK2V617F mutations occur decades before
BCR::ABL1-negative MPN diagnosis, increase the fitness of HSCs, and induce a megakaryocyte–erythroid differentiation bias [
67]. CHIP/ARCH individuals also have an increased risk of developing hematologic malignancies or cardiovascular diseases in patients with additional mutations [
68].
Some studies have investigated the impact of genetic background on the risk of developing
BCR::ABL1-negative MPNs. For example, the
JAK2 46/1 haplotype concomitant with
JAK2V617F,
RBBP6,
SH2B3, and
TERT mutations was shown to increase the risk of developing
BCR::ABL1-negative MPNs [
69,
70,
71,
72,
73,
74]. A GWAS analysis of 888,503 individuals, including 2949 patients with
BCR::ABL1-negative MPNs, identified 17 loci, including
JAK2 and
TERT [
18]. SNPs located at the identified loci, such as rs17879961 (located at
CHEK2 exon 5) and rs534137 (located at the promoter region of
GFI1B), were considered to induce the instability of HSCs homeostasis and may predispose patients to
BCR::ABL1-negative MPNs.
In addition to SNPs that confer higher susceptibility to develop
BCR::ABL1-negative MPNs, SNPs that affect the phenotype, prognosis, and response to therapies of
BCR::ABL1-negative MPNs have been reported and summarized [
75]. Although allelic frequencies are not rare (0.114822 for rs6198, 0.279039 for rs1024611, and 0.444187 for rs2431697 by gnomAD, respectively), a poorer prognosis was observed among patients with PMF harboring both
JAK2V617F and homozygous mutations of rs6198 locating at
NR3C1 than those bearing wild-type
NR3C1 [
76], rs1024611 at
CCL2 strongly correlated to the
CCL2 expression and the myelofibrosis grade [
77], and homozygous rs2431697 at miR-146a was associated with myelofibrosis progression [
78].