Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | George E. Zakynthinos | -- | 1805 | 2023-10-18 22:44:05 | | | |
| 2 | Catherine Yang | Meta information modification | 1805 | 2023-10-19 03:09:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zakynthinos, G.E.; Tsolaki, V.; Oikonomou, E.; Vavouranakis, M.; Siasos, G.; Zakynthinos, E. Metabolic Syndrome and Atrial Fibrillation. Encyclopedia. Available online: https://encyclopedia.pub/entry/50490 (accessed on 25 July 2026).
Zakynthinos GE, Tsolaki V, Oikonomou E, Vavouranakis M, Siasos G, Zakynthinos E. Metabolic Syndrome and Atrial Fibrillation. Encyclopedia. Available at: https://encyclopedia.pub/entry/50490. Accessed July 25, 2026.
Zakynthinos, George E., Vasiliki Tsolaki, Evangelos Oikonomou, Manolis Vavouranakis, Gerasimos Siasos, Epaminondas Zakynthinos. "Metabolic Syndrome and Atrial Fibrillation" Encyclopedia, https://encyclopedia.pub/entry/50490 (accessed July 25, 2026).
Zakynthinos, G.E., Tsolaki, V., Oikonomou, E., Vavouranakis, M., Siasos, G., & Zakynthinos, E. (2023, October 18). Metabolic Syndrome and Atrial Fibrillation. In Encyclopedia. https://encyclopedia.pub/entry/50490
Zakynthinos, George E., et al. "Metabolic Syndrome and Atrial Fibrillation." Encyclopedia. Web. 18 October, 2023.
Copy Citation
Obesity, hypertension, insulin resistance, and dyslipidemia are all clusters of an entity called “Metabolic Syndrome”. The global trends of this syndrome’s incidence/prevalence continue to increase reciprocally, converting it into a massive epidemic problem in the medical community.
atrial fibrillation
metabolic syndrome
obesity
1. Obesity
Atrial fibrillation is markedly more prevalent among obese individuals compared to those with a BMI below 30.0 kg/m2 [1][2]. Remarkably, even within the latter group, an elevated level of abdominal fat has been associated with an increased risk of AF occurrence [2]. Notably, an expanded body size during early adulthood, elevated BMI in midlife, and weight gain from the age of 20 to midlife have all been linked to an augmented likelihood of future AF development. This highlights that increased BMI at any stage of life can potentially serve as a risk factor for AF [3]. Individuals classified as obese face nearly a 50% heightened risk of developing AF when compared to their non-obese counterparts [4].
Obesity exerts a twofold influence by altering both cardiac hemodynamics and heart morphology. This leads to an amplified blood volume, increased cardiac output, remodeling of cardiac chambers, and an augmentation of epicardial fat [5]. Cardiac remodeling precipitated by obesity encompasses the emergence of left ventricular hypertrophy, resulting in diastolic dysfunction and pronounced enlargement of the left atrium [5][6]. These effects, in conjunction with the altered hemodynamics—such as heightened stroke volume and pulmonary pressures—establish an environment conducive to the initiation of AF [6]. The potential for echocardiographic left atrial volumetric enlargement over a decade is almost 2.4 times higher in obese individuals [7]. Moreover, pericardial adipose tissue significantly contributes to the burgeoning frequency of AF [8]. Of particular note, epicardial fat secretes adipocytokines, including Activin-1, which incites fibroblast proliferation and amplifies fibrosis within myocardial tissue [9]. This phenomenon plays a role in shaping the substrate required for AF.
Mahajan et al. highlighted the impact of obesity on cardiac tissue in sheep, demonstrating electroanatomical changes. Significantly reduced mean conduction velocity was observed in the obese group (LA 1.18 ± 0.04 m/s vs. 1.58 ± 0.04 m/s, p < 0.001) compared to the normal group. While the mean total voltage remained unchanged, distinct regional voltage patterns emerged in the left atrium of the obese and control groups, primarily due to a substantial reduction in posterior LA voltage (3.7 ± 2.3 mV vs. 5.5 ± 2.3 mV; p < 0.001) [10]. These findings were recently corroborated in men by the same research group [conduction velocity: 0.86 ± 0.31 m/s vs. 1.26 ± 0.29 m/s; p < 0.001]. In the obese group, 13.9% of all points in the left atrium exhibited low voltage, compared to 3.4% in the reference group (p < 0.001) [11]. These electrical changes contribute to the creation and maintenance of the substrate required for AF occurrence.
2. Hypertension
Hypertension contributes to around 14% of all cases of AF, and among AF patients, over 70% have hypertension. Hypertension independently amplifies the risk of AF progression and leads to the emergence of adverse effects associated with AF [12]. The impact of long-standing hypertension on the heart is well-documented, resulting in elevated cardiac filling pressures and diastolic dysfunction. A study from Norway established a link between diastolic dysfunction and AF [13]. Notably, this connection is applicable in cases with both left atrial enlargement and without it, where elevated left ventricular filling pressures primarily drive AF [14]. However, the most significant structural alteration attributed to hypertension is the enlargement of the left atrium (LA), influenced by various mechanisms encompassing hemodynamic and electrical remodeling, neurohormonal activation, and inflammation [15]. Electrical remodeling is also evident in individuals with hypertension, characterized by extensive areas of double potentials, fractionated signals, and varying and slower regional activation times within the atria, leading to atrial conduction delays [16]. Another noteworthy aspect of hypertension is the role of the renin-angiotensin-aldosterone system (RAAS), which becomes highly activated in hypertensive individuals. Experiments conducted on mice revealed that elevated levels of angiotensin II trigger both electrophysiological and structural modifications in the heart, along with increased fibrosis in the left atrium [17][18]. This fibrosis, as mentioned earlier, constitutes a significant factor in the development of AF [19]. Moreover, an alternate pathway through which hypertension may lead to fibrosis and subsequently to AF involves the extensive activation of the inflammatory process [20].
3. Insulin Resistance/Diabetes
Individuals with either type of diabetes have a twofold higher prevalence of AF, a figure that steadily rises as the severity of the disease and its microvascular complications progress. The presence of silent AF episodes is highly probable due to autonomic dysfunction [21]. Similar to hypertension, diabetes also involves structural and electrical remodeling, autonomic dysregulation, and inflammation. The renin–angiotensin–aldosterone system (RAAS) is enhanced, and elevated levels of angiotensin II are linked to diabetes, contributing to atrial fibrosis [22]. Another contributing factor in diabetes is the increased production of advanced glycation end products (AGEs) and their receptors, potentially exacerbating atrial scarring and fibrosis and thus contributing to the substrate for AF [23]. Consequently, patients develop diabetic cardiomyopathy characterized by diastolic dysfunction, which creates conditions within the atria conducive to AF occurrence [24]. Diabetic patients exhibit delayed conduction velocities and atrial emptying [25], intra-atrial electromechanical delay [26], prolonged action potentials, and abnormal atrial voltage [27]. These observations might be explained by the pathological expression of gap-junction proteins known as connexins [28]. Autonomic abnormalities, including sympathetic upregulation leading to sympathetic denervation and an imbalance in the autonomic nervous system—termed diabetic neuropathy—also impact the heart and can contribute to AF [29][30]. Inflammation and oxidative stress further contribute to the proarrhythmic state of diabetes. Disturbed mitochondrial function prevents the elimination of reactive oxygen species (ROS), leading to the activation of inflammation pathways evidenced by elevated inflammatory markers [31]. Additionally, hypoglycemia, with its accompanying sympathetic activation and fluctuations in blood glucose levels, can reinforce myocardial fibrosis and elevate oxidative stress, forming unique key mediators of the arrhythmic substrate in diabetes [32][33].
4. Dyslipidemia
The role of dyslipidemia in the occurrence of AF has not yet been fully elucidated. Low HDL cholesterol has been associated with an increased risk of AF, although elevated triglycerides have not shown a similar correlation [34]. However, findings from a Japanese cohort indicated that the relationship between low HDL and AF risk exists only in women, not men [35]. In contrast to the preceding clusters of MetS, the underlying mechanisms connecting dyslipidemia to AF have not been extensively investigated. Elevated blood lipids create an inflammatory environment and increase oxidative stress [36], potentially contributing to AF development. A comparison between patients with and without AF revealed that the AF group had 1.6-fold higher plasma triglycerides and increased inflammation markers [37]. Increased myocardial triglyceride content (MTGC), as measured by magnetic resonance spectroscopy, has been linked to diastolic dysfunction, although the precise connection between MTGC and plasma triglycerides remains unclear [38]. Recent studies have suggested a connection between postprandial, very low-density lipoprotein (VLDL), composed of triglycerides, and atrial remodeling in MetS patients [39]. Further support for this observation comes from new data demonstrating that a surplus of triglycerides within VLDL leads to atrial enlargement and disturbances in PR duration. In MetS patients, the left atrium diameter and volume were larger compared to non-MetS individuals (LA diameter: non-MetS 3.2 ± 0.3 cm vs. MetS-off statin 4.4 ± 0.4 cm vs. MetS-on statin 4.3 ± 0.3 cm, p < 0.0001; LA maximum volume: non-MetS 45.2 ± 9.5 mL vs. MetS-off statin 81.9 ± 13.9 mL vs. MetS-on statin 77.5 ± 18.9 mL, p < 0.0001; LA minimum volume: non-MetS 28.1 ± 7.5 mL vs. MetS-off statin 39.3 ± 10.8 mL vs. MetS-on statin 42.0 ± 11.3 mL, p = 0.0065). Additionally, atrioventricular conduction, as measured by PR interval, was prolonged in MetS patients (176.1 ± 19.0 ms vs. 156.2 ± 15.4 ms, p = 0.0014) [40]. Therefore, it is plausible that dyslipidemia contributes to structural and electrical changes in the cardiac chambers, yet further research is warranted in this area.
5. Metabolic Syndrome
It is readily apparent that when these risk factors converge within the framework of “Metabolic syndrome,” they profoundly disrupt the proper functioning of the left atrium, precipitating the occurrence of AF through a multitude of mechanisms. Overall, there is insufficient evidence to substantiate the direct association of MetS as a distinct entity with AF. Instead, much of the research has focused on the individual components of MetS in relation to AF. A rat study revealed that obesity compounds the atrial arrhythmogenic phenotype in hypertensive rats, exacerbating interstitial atrial fibrotic changes, conduction velocities, and left atrial emptying [41]. This lends credence to the idea that MetS, as an integrated entity, can indeed instigate AF. Additionally, evidence suggests that MetS patients experience both structural alterations—such as left ventricular and left atrial remodeling—and electrical changes (PR interval in MetS group vs. non-MetS: 167.6 ± 20.0 msec vs. 156.2 ± 24.9 msec, p = 0.0064) [39][40]. Furthermore, both MetS and AF are characterized by inflammation, and their coexistence has been associated with even greater levels of inflammation, potentially introducing another mechanism that leads to fibrosis and AF development [42]. Beyond inflammatory markers, other biomarkers dysregulated in MetS—such as adiponectin, leptin, ghrelin, uric acid, and OxLDL—appear to contribute to the initiation and progression of AF. Adiponectin appears to mitigate cardiac chamber remodeling [43]. Leptin-mediated pathways impact Angiotensin metabolism, thus contributing to AF [44]. Moreover, since leptin regulates calcium homeostasis, it significantly influences electrophysiological pathways [45]. Lastly, MetS disrupts autonomic tone, with the degree of impairment corresponding to the number of MetS clusters [46]. This observation suggests that MetS could potentially play a role not only in the formation of substrate but also in triggering AF (Figure 1).
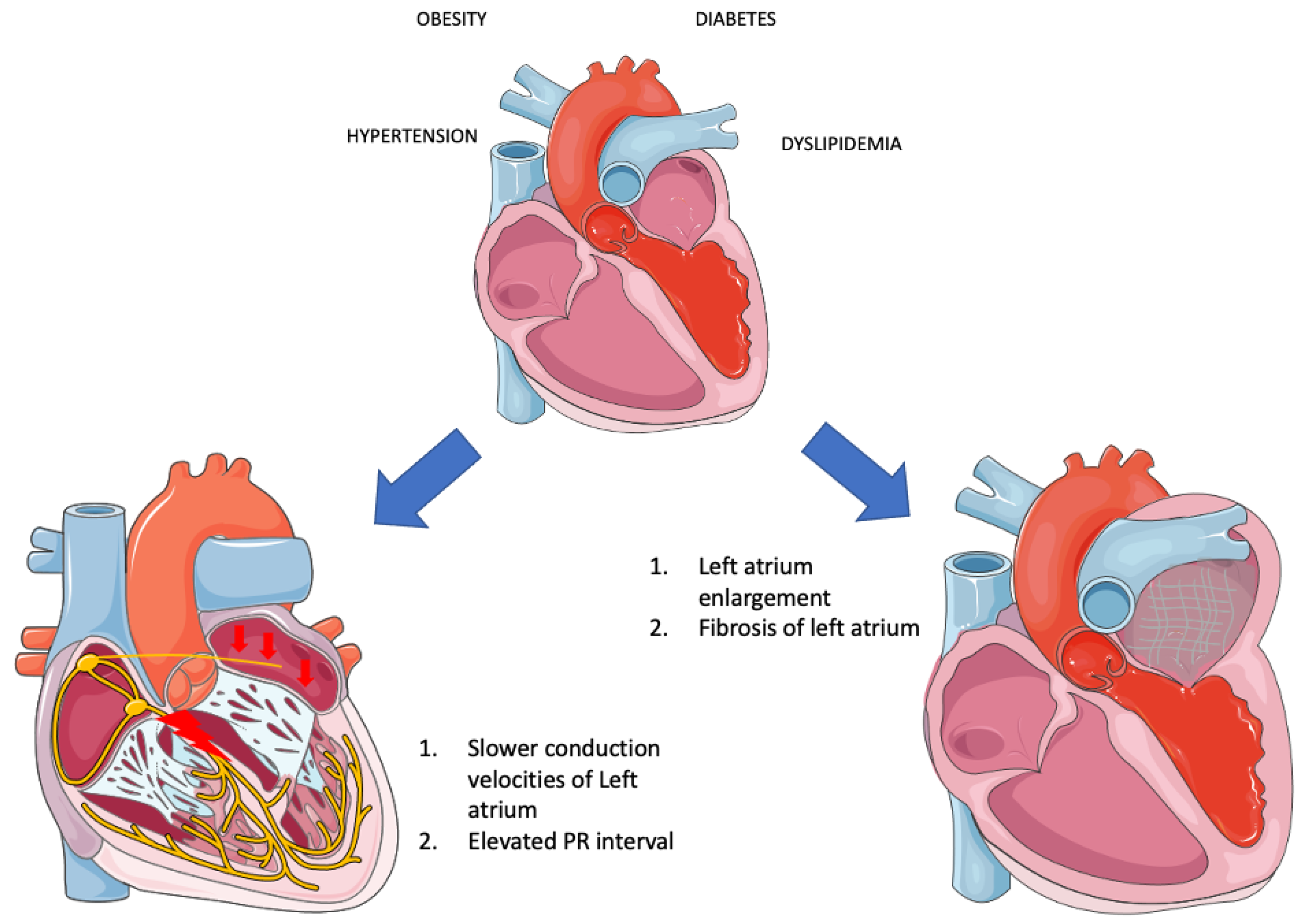
Figure 1. Electrical and structural remodeling in metabolic syndrome (Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/, accessed on 20 August 2023).
The human heart, alongside skeletal muscles, kidneys, and the brain, is a highly energy-consuming organ, necessitating significant amounts of adenosine triphosphate (ATP) molecules for proper function. Approximately 60% of the required energy is derived from fatty acid (FA) metabolism through β-oxidation [47]. However, this process is accompanied by a reduction in the NADH/NAD+ system within mitochondria, leading to the generation of electrons that enter the electron transport chain (ETC) to produce ATP. In situations where β-oxidation becomes overactive, or ATP levels decrease, an excess of electrons is generated, resulting in the conversion of these electrons into superoxide radicals (ROS) [48]. Metabolic syndrome has been demonstrated to lead to a condition in which the heart predominantly relies on FA for ATP production, imposing excessive stress on mitochondria and causing damage through various mechanisms [49]. The malfunctioning ATP production mechanisms lead to a decreased ATP-to-O2 consumption ratio, triggering hyperactivity of the cardiac muscle, subsequent hypertrophy, and diastolic dysfunction. Moreover, mitochondria play a crucial role in maintaining Ca2+ homeostasis. Thus, mitochondrial dysfunction results in disruptions and oscillations of intracellular Ca2+ levels, potentially inducing arrhythmias [50]. Lastly, due to the incapacity of mitochondria to generate ATP and their shift towards ROS production in the ETC, substantial quantities of ROS are released into circulation [49]. As mentioned earlier, ROS triggers inflammation, leading to structural remodeling of the heart as previously described. Consequently, recent findings underscore the significance of mitophagy—the process that selectively eliminates damaged mitochondria from cells, facilitating their degradation by lysosomes and maintaining mitochondrial homeostasis—in the pathophysiology of Metabolic Syndrome (MetS). There is a suggestion for its potential therapeutic application in the future [51].
References
- Wang, T.J.; Parise, H.; Levy, D.; D’Agostino, R.B., Sr.; Wolf, P.A.; Vasan, R.S.; Benjamin, E.J. Obesity and the risk of new-onset atrial fibrillation. JAMA 2004, 292, 2471–2477.
- Baek, Y.; Yang, P.; Kim, T.; Uhm, J.; Park, J.; Pak, H.; Lee, M.; Joung, B. Associations of Abdominal Obesity and New-Onset Atrial Fibrillation in the General Population. J. Am. Heart Assoc. 2017, 6, e004705.
- Rosengren, A.; Hauptman, P.J.; Lappas, G.; Olsson, L.; Wilhelmsen, L.; Swedberg, K. Big men and atrial fibrillation: Effects of body size and weight gain on risk of atrial fibrillation in men. Eur. Heart J. 2009, 30, 1113–1120.
- Wanahita, N.; Messerli, F.H.; Bangalore, S.; Gami, A.S.; Somers, V.K.; Steinberg, J.S. Atrial fibrillation and obesity—Results of a meta-analysis. Am. Heart J. 2008, 155, 310–315.
- Alpert, M.A.; Omran, J.; Bostick, B.P. Effects of Obesity on Cardiovascular Hemodynamics, Cardiac Morphology, and Ventricular Function. Curr. Obes. Rep. 2016, 5, 424–434.
- Lavie, C.J.; Pandey, A.; Lau, D.H.; Alpert, M.A.; Sanders, P. Obesity and Atrial Fibrillation Prevalence, Pathogenesis, and Prognosis: Effects of Weight Loss and Exercise. J. Am. Coll. Cardiol. 2017, 70, 2022–2035.
- Magnani, J.W.; Hylek, E.M.; Apovian, C.M. Obesity begets atrial fibrillation: A contemporary summary. Circulation 2013, 128, 401–405.
- Hatem, S.N.; Sanders, P. Epicardial adipose tissue and atrial fibrillation. Cardiovasc. Res. 2014, 102, 205–213.
- Venteclef, N.; Guglielmi, V.; Balse, E.; Gaborit, B.; Cotillard, A.; Atassi, F.; Amour, J.; Leprince, P.; Dutour, A.; Clément, K.; et al. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur. Heart J. 2015, 36, 795–805.
- Mahajan, R.; Lau, D.H.; Brooks, A.G.; Shipp, N.J.; Manavis, J.; Wood, J.P.; Finnie, J.W.; Samuel, C.S.; Royce, S.G.; Twomey, D.J.; et al. Electrophysiological, Electroanatomical, and Structural Remodeling of the Atria as Consequences of Sustained Obesity. J. Am. Coll. Cardiol. 2015, 66, 1–11.
- Mahajan, R.; Nelson, A.; Pathak, R.K.; Middeldorp, M.E.; Wong, C.X.; Twomey, D.J.; Carbone, A.; Teo, K.; Agbaedeng, T.; Linz, D.; et al. Electroanatomical Remodeling of the Atria in Obesity: Impact of Adjacent Epicardial Fat. JACC Clin. Electrophysiol. 2018, 4, 1529–1540.
- Lip, G.Y.H.; Coca, A.; Kahan, T.; Boriani, G.; Manolis, A.S.; Olsen, M.H.; Oto, A.; Potpara, T.S.; Steffel, J.; Marín, F.; et al. Hypertension and cardiac arrhythmias: A consensus document from the European Heart Rhythm Association (EHRA) and ESC Council on Hypertension, endorsed by the Heart Rhythm Society (HRS), Asia-Pacific Heart Rhythm Society (APHRS) and Sociedad Latinoamericana de Estimulación Cardíaca y Electrofisiología (SOLEACE). Europace 2017, 19, 891–911.
- Tiwari, S.; Schirmer, H.; Jacobsen, B.K.; Hopstock, L.; Nyrnes, A.; Heggelund, G.; Njølstad, I.; Mathiesen, E.B.; Løchen, M.-L. Association between diastolic dysfunction and future atrial fibrillation in the Tromsø Study from 1994 to 2010. Heart 2015, 101, 1302–1308.
- Takagi, T.; Takagi, A.; Yoshikawa, J. Elevated left ventricular filling pressure estimated by E/E′ ratio after exercise predicts development of new-onset atrial fibrillation independently of left atrial enlargement among elderly patients without obvious myocardial ischemia. J. Cardiol. 2014, 63, 128–133.
- Kockskämper, J.; Pluteanu, F. Left Atrial Myocardium in Arterial Hypertension. Cells 2022, 11, 3157.
- Medi, C.; Kalman, J.M.; Spence, S.J.; Teh, A.W.; Lee, G.; Bader, I.; Kaye, D.M.; Kistler, P.M. Atrial Electrical and Structural Changes Associated with Longstanding Hypertension in Humans: Implications for the Substrate for Atrial Fibrillation. J. Cardiovasc. Electrophysiol. 2011, 22, 1317–1324.
- Jansen, H.J.; Mackasey, M.; Moghtadaei, M.; Liu, Y.; Kaur, J.; Egom, E.E.; Tuomi, J.M.; Rafferty, S.A.; Kirkby, A.W.; Rose, R.A. NPR-C (Natriuretic Peptide Receptor-C) Modulates the Progression of Angiotensin II–Mediated Atrial Fibrillation and Atrial Remodeling in Mice. Circ. Arrhythmia Electrophysiol. 2019, 12, e006863.
- Jansen, H.J.; Mackasey, M.; Moghtadaei, M.; Belke, D.D.; Egom, E.E.; Tuomi, J.M.; Rafferty, S.A.; Kirkby, A.W.; Rose, R.A. Distinct patterns of atrial electrical and structural remodeling in angiotensin II mediated atrial fibrillation. J. Mol. Cell. Cardiol. 2018, 124, 12–25.
- Lau, D.H.; Mackenzie, L.; Kelly, D.J.; Psaltis, P.J.; Brooks, A.G.; Worthington, M.; Rajendram, A.; Kelly, D.R.; Zhang, Y.; Kuklik, P.; et al. Hypertension and atrial fibrillation: Evidence of progressive atrial remodeling with electrostructural correlate in a conscious chronically instrumented ovine model. Heart Rhythm. 2010, 7, 1282–1290.
- Yao, Y.; Yang, M.; Liu, D.; Zhao, Q. Immune remodeling and atrial fibrillation. Front. Physiol. 2022, 13, 927221.
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498, Erratum in Eur. Heart J. 2021, 42, 507, Erratum in Eur. Heart J. 2021, 42, 546–547, Erratum in Eur. Heart J. 2021, 42, 4194.
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2015, 90, 84–93.
- Kato, T.; Yamashita, T.; Sekiguchi, A.; Tsuneda, T.; Sagara, K.; Takamura, M.; Kaneko, S.; Aizawa, T.; Fu, L.-T. AGEs-RAGE System Mediates Atrial Structural Remodeling in the Diabetic Rat. J. Cardiovasc. Electrophysiol. 2008, 19, 415–420.
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic cardiomyopathy: A hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 2017, 61, 21–28.
- Ayhan, S.; Ozturk, S.; Alcelik, A.; Ozlu, M.F.; Erdem, A.; Memioglu, T.; Ozdemir, M.; Yazici, M. Atrial conduction time and atrial mechanical function in patients with impaired fasting glucose. J. Interv. Card. Electrophysiol. 2012, 35, 247–252.
- Demir, K.; Avci, A.; Kaya, Z.; Marakoglu, K.; Ceylan, E.; Yilmaz, A.; Ersecgin, A.; Armutlukuyu, M.; Altunkeser, B.B. Assessment of atrial electromechanical delay and P-wave dispersion in patients with type 2 diabetes mellitus. J. Cardiol. 2016, 67, 378–383.
- Chao, T.-F.; Suenari, K.; Chang, S.-L.; Lin, Y.-J.; Lo, L.-W.; Hu, Y.-F.; Tuan, T.-C.; Tai, C.-T.; Tsao, H.-M.; Li, C.-H.; et al. Atrial Substrate Properties and Outcome of Catheter Ablation in Patients with Paroxysmal Atrial Fibrillation Associated With Diabetes Mellitus or Impaired Fasting Glucose. Am. J. Cardiol. 2010, 106, 1615–1620.
- Watanabe, M.; Yokoshiki, H.; Mitsuyama, H.; Mizukami, K.; Ono, T.; Tsutsui, H. Conduction and refractory disorders in the diabetic atrium. Am. J. Physiol. Circ. Physiol. 2012, 303, H86–H95.
- Kuehl, M.; Stevens, M.J. Cardiovascular autonomic neuropathies as complications of diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 405–416.
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898.
- Faria, A.; Persaud, S.J. Cardiac oxidative stress in diabetes: Mechanisms and therapeutic potential. Pharmacol. Ther. 2017, 172, 50–62.
- Ko, S.-H.; Park, Y.-M.; Yun, J.-S.; Cha, S.-A.; Choi, E.-K.; Han, K.; Han, E.; Lee, Y.-H.; Ahn, Y.-B. Severe hypoglycemia is a risk factor for atrial fibrillation in type 2 diabetes mellitus: Nationwide population-based cohort study. J. Diabetes Complicat. 2018, 32, 157–163.
- Saito, S.; Teshima, Y.; Fukui, A.; Kondo, H.; Nishio, S.; Nakagawa, M.; Saikawa, T.; Takahashi, N. Glucose fluctuations increase the incidence of atrial fibrillation in diabetic rats. Cardiovasc. Res. 2014, 104, 5–14.
- Zheng, Y.; Xie, Z.; Li, J.; Chen, C.; Cai, W.; Dong, Y.; Xue, R.; Liu, C. Meta-analysis of metabolic syndrome and its individual components with risk of atrial fibrillation in different populations. BMC Cardiovasc. Disord. 2021, 21, 90.
- Watanabe, H.; Tanabe, N.; Yagihara, N.; Watanabe, T.; Aizawa, Y.; Kodama, M. Association Between Lipid Profile and Risk of Atrial Fibrillation. Circ. J. 2011, 75, 2767–2774.
- Siasos, G.; Tousoulis, D.; Oikonomou, E.; Zaromitidou, M.; Stefanadis, C.; Papavassiliou, A.G. Inflammatory Markers in Hyperlipidemia: From Experimental Models to Clinical Practice. Curr. Pharm. Des. 2011, 17, 4132–4146.
- Kim, S.-M.; Kim, J.-M.; Shin, D.-G.; Kim, J.-R.; Cho, K.-H. Relation of atrial fibrillation (AF) and change of lipoproteins: Male patients with AF exhibited severe pro-inflammatory and pro-atherogenic properties in lipoproteins. Clin. Biochem. 2014, 47, 869–875.
- Soghomonian, A.; Dutour, A.; Kachenoura, N.; Thuny, F.; Lasbleiz, A.; Ancel, P.; Cristofari, R.; Jouve, E.; Simeoni, U.; Kober, F.; et al. Is increased myocardial triglyceride content associated with early changes in left ventricular function? A 1H-MRS and MRI strain study. Front. Endocrinol. 2023, 14, 1181452.
- Lee, H.-C.; Shin, S.-J.; Huang, J.-K.; Lin, M.-Y.; Lin, Y.-H.; Ke, L.-Y.; Jiang, H.-J.; Tsai, W.-C.; Chao, M.-F.; Lin, Y.-H. The role of postprandial very-low-density lipoprotein in the development of atrial remodeling in metabolic syndrome. Lipids Health Dis. 2020, 19, 210.
- Lee, H.-C.; Cheng, W.-C.; Ma, W.-L.; Lin, Y.-H.; Shin, S.-J.; Lin, Y.-H. Association of lipid composition and unsaturated fatty acids of VLDL with atrial remodeling in metabolic syndrome. Sci. Rep. 2023, 13, 6575.
- Hohl, M.; Lau, D.H.; Müller, A.; Elliott, A.D.; Linz, B.; Mahajan, R.; Hendriks, J.M.L.; Böhm, M.; Schotten, U.; Sanders, P.; et al. Concomitant Obesity and Metabolic Syndrome Add to the Atrial Arrhythmogenic Phenotype in Male Hypertensive Rats. J. Am. Heart Assoc. 2017, 6, e006717.
- Itani, H.A.; Jaffa, M.A.; Elias, J.; Sabra, M.; Zakka, P.; Ballout, J.; Bekdash, A.; Ibrahim, R.; Al Hariri, M.; Ghemrawi, M.; et al. Genomic and Proteomic Study of the Inflammatory Pathway in Patients with Atrial Fibrillation and Cardiometabolic Syndrome. Front. Cardiovasc. Med. 2020, 7, 613271.
- Rafaqat, S.; Sharif, S.; Majeed, M.; Naz, S.; Manzoor, F.; Rafaqat, S. Biomarkers of Metabolic Syndrome: Role in Pathogenesis and Pathophysiology of Atrial Fibrillation. J. Atr. Fibrillation 2021, 14, 20200495.
- Fukui, A.; Takahashi, N.; Nakada, C.; Masaki, T.; Kume, O.; Shinohara, T.; Teshima, Y.; Hara, M.; Saikawa, T. Role of Leptin Signaling in the Pathogenesis of Angiotensin II–Mediated Atrial Fibrosis and Fibrillation. Circ. Arrhythmia Electrophysiol. 2013, 6, 402–409.
- Lin, Y.-K.; Chen, Y.-C.; Chang, S.-L.; Lin, Y.-J.; Chen, J.-H.; Yeh, Y.-H.; Chen, S.-A.; Chen, Y.-J. Heart failure epicardial fat increases atrial arrhythmogenesis. Int. J. Cardiol. 2013, 167, 1979–1983.
- Gehi, A.K.; Lampert, R.; Veledar, E.; Lee, F.; Goldberg, J.; Jones, L.; Murrah, N.; Ashraf, A.; Vaccarino, V. A Twin Study of Metabolic Syndrome and Autonomic Tone. J. Cardiovasc. Electrophysiol. 2009, 20, 422–428.
- Li, J.; Li, J.; Chen, Y.; Hu, W.; Gong, X.; Qiu, H.; Chen, H.; Xin, Y.; Li, H. The Role of Mitochondria in Metabolic Syndrome–Associated Cardiomyopathy. Oxidative Med. Cell. Longev. 2022, 2022, 9196232.
- Panov, A.; Mayorov, V.I.; Dikalov, S. Metabolic Syndrome and β-Oxidation of Long-Chain Fatty Acids in the Brain, Heart, and Kidney Mitochondria. Int. J. Mol. Sci. 2022, 23, 4047.
- Federico, M.; De la Fuente, S.; Palomeque, J.; Sheu, S. The role of mitochondria in metabolic disease: A special emphasis on heart dysfunction. J. Physiol. 2021, 599, 3477–3493.
- Sommese, L.; Valverde, C.A.; Blanco, P.; Castro, M.C.; Rueda, O.V.; Kaetzel, M.; Dedman, J.; Anderson, M.E.; Mattiazzi, A.; Palomeque, J. Ryanodine receptor phosphorylation by CaMKII promotes spontaneous Ca2+ release events in a rodent model of early stage diabetes: The arrhythmogenic substrate. Int. J. Cardiol. 2016, 202, 394–406.
- Miao, M.; Han, Y.; Liu, L. Mitophagy in metabolic syndrome. J. Clin. Hypertens. 2023, 25, 397–403.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
701
Revisions:
2 times
(View History)
Update Date:
19 Oct 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No