 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sungho Maeng | -- | 3980 | 2023-10-15 08:50:20 | | | |
| 2 | Jessie Wu | + 5 word(s) | 3985 | 2023-10-16 04:29:32 | | |
Video Upload Options
Alzheimer’s disease (AD) is a neurodegenerative disorder associated with cognitive decline. Despite worldwide efforts to find a cure, no proper treatment has been developed yet, and the only effective countermeasure is to prevent the disease progression by early diagnosis. The reason why new drug candidates fail to show therapeutic effects in clinical studies may be due to misunderstanding the cause of AD. Regarding the cause of AD, the most widely known is the amyloid cascade hypothesis, in which the deposition of amyloid beta and hyperphosphorylated tau is the cause. However, many new hypotheses were suggested. Among them, based on preclinical and clinical evidence supporting a connection between AD and diabetes, insulin resistance has been pointed out as an important factor in the development of AD.
1. Insulin Resistance Is Linked to Alzheimer’s Disease
1.1. Brain Energy Mobilization
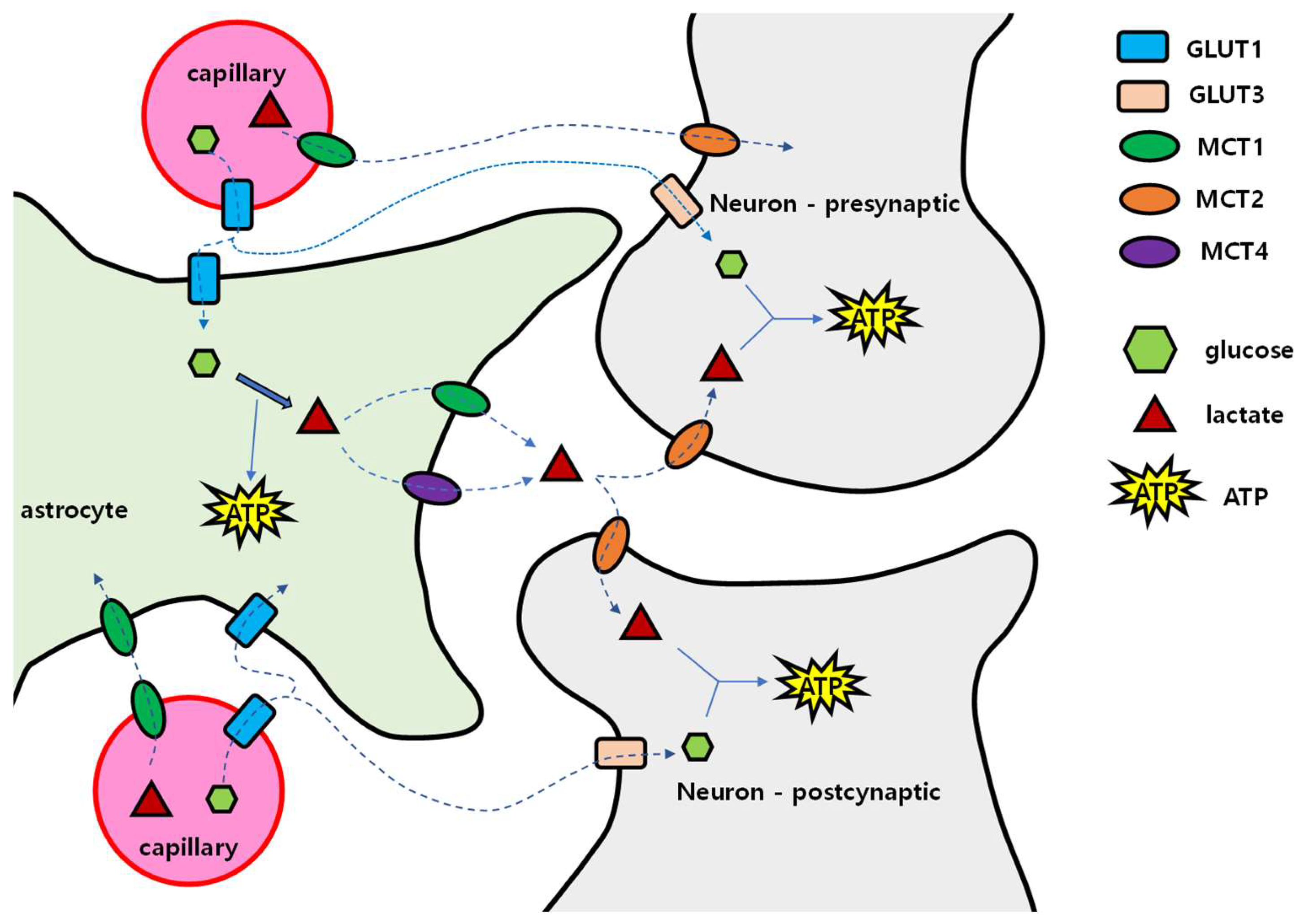
1.2. When and How the Brain Deals with the Lack of Glucose
2. How Insulin Resistance Causes Dementia
2.1. Brain Glucose Insufficiency
2.2. Brain Insulin Insufficiency
References
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160.
- Tuligenga, R.H.; Dugravot, A.; Tabak, A.G.; Elbaz, A.; Brunner, E.J.; Kivimaki, M.; Singh-Manoux, A. Midlife type 2 diabetes and poor glycaemic control as risk factors for cognitive decline in early old age: A post-hoc analysis of the Whitehall II cohort study. Lancet Diabetes Endocrinol. 2014, 2, 228–235.
- Sokoloff, L. Energetics of functional activation in neural tissues. Neurochem. Res. 1999, 24, 321–329.
- Yu, L.; Yu, Y. Energy-efficient neural information processing in individual neurons and neuronal networks. J. Neurosci. Res. 2017, 95, 2253–2266.
- Lying-Tunell, U.; Lindblad, B.S.; Malmlund, H.O.; Persson, B. Cerebral blood flow and metabolic rate of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids. Acta Neurol. Scand. 1980, 62, 265–275.
- Holliday, M.A. Metabolic rate and organ size during growth from infancy to maturity and during late gastation and early infancy. Pediatrics 1971, 47 (Suppl. S2), 169.
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch. 2020, 472, 1299–1343.
- Lebovitz, H.E. Insulin resistance: Definition and consequences. Exp. Clin. Endocrinol. Diabetes 2001, 109 (Suppl. S2), S135–S148.
- Leybaert, L.; De Bock, M.; Van Moorhem, M.; Decrock, E.; De Vuyst, E. Neurobarrier coupling in the brain: Adjusting glucose entry with demand. J. Neurosci. Res. 2007, 85, 3213–3220.
- Hargreaves, M.; Spriet, L.L. Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020, 2, 817–828.
- Bergersen, L.H. Is lactate food for neurons? Comparison of monocarboxylate transporter subtypes in brain and muscle. Neuroscience 2007, 145, 11–19.
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessieres, B.; Gao, V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018, 66, 1244–1262.
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823.
- Morris, A.A. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005, 28, 109–121.
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22.
- Wilkins, J.M.; Trushina, E. Application of Metabolomics in Alzheimer’s Disease. Front. Neurol. 2017, 8, 719.
- Chassard, C.; Scott, K.P.; Marquet, P.; Martin, J.C.; Del’homme, C.; Dapoigny, M.; Flint, H.J.; Bernalier-Donadille, A. Assessment of metabolic diversity within the intestinal microbiota from healthy humans using combined molecular and cultural approaches. FEMS Microbiol. Ecol. 2008, 66, 496–504.
- Bugaut, M. Occurrence, absorption and metabolism of short chain fatty acids in the digestive tract of mammals. Comp. Biochem. Physiol. B 1987, 86, 439–472.
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478.
- Rhea, E.M.; Banks, W.A. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front. Neurosci. 2019, 13, 521.
- Newington, J.T.; Harris, R.A.; Cumming, R.C. Reevaluating Metabolism in Alzheimer’s Disease from the Perspective of the Astrocyte-Neuron Lactate Shuttle Model. J. Neurodegener. Dis. 2013, 2013, 234572.
- Rhea, E.M.; Salameh, T.S.; Banks, W.A. Routes for the delivery of insulin to the central nervous system: A comparative review. Exp. Neurol. 2019, 313, 10–15.
- Seaquist, E.R.; Tkac, I.; Damberg, G.; Thomas, W.; Gruetter, R. Brain glucose concentrations in poorly controlled diabetes mellitus as measured by high-field magnetic resonance spectroscopy. Metabolism 2005, 54, 1008–1013.
- Berthet, C.; Lei, H.; Thevenet, J.; Gruetter, R.; Magistretti, P.J.; Hirt, L. Neuroprotective role of lactate after cerebral ischemia. J. Cereb. Blood Flow. Metab. 2009, 29, 1780–1789.
- Schousboe, A.; Westergaard, N.; Sonnewald, U.; Petersen, S.B.; Huang, R.; Peng, L.; Hertz, L. Glutamate and glutamine metabolism and compartmentation in astrocytes. Dev. Neurosci. 1993, 15, 359–366.
- Danysz, W.; Parsons, C.G. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer’s disease: Preclinical evidence. Int. J. Geriatr. Psychiatry 2003, 18 (Suppl. S1), S23–S32.
- Souza, D.G.; Almeida, R.F.; Souza, D.O.; Zimmer, E.R. The astrocyte biochemistry. Semin. Cell. Dev. Biol. 2019, 95, 142–150.
- Liu, D.; Ke, Z.; Luo, J. Thiamine Deficiency and Neurodegeneration: The Interplay Among Oxidative Stress, Endoplasmic Reticulum Stress, and Autophagy. Mol. Neurobiol. 2017, 54, 5440–5448.
- Alexander-Kaufman, K.; Harper, C. Transketolase: Observations in alcohol-related brain damage research. Int. J. Biochem. Cell. Biol. 2009, 41, 717–720.
- Green, P.H. Alcohol, nutrition and malabsorption. Clin. Gastroenterol. 1983, 12, 563–574.
- Karuppagounder, S.S.; Xu, H.; Shi, Q.; Chen, L.H.; Pedrini, S.; Pechman, D.; Baker, H.; Beal, M.F.; Gandy, S.E.; Gibson, G.E. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer’s mouse model. Neurobiol. Aging 2009, 30, 1587–1600.
- Zhang, Q.; Yang, G.; Li, W.; Fan, Z.; Sun, A.; Luo, J.; Ke, Z.J. Thiamine deficiency increases beta-secretase activity and accumulation of beta-amyloid peptides. Neurobiol. Aging 2011, 32, 42–53.
- Zubaran, C.; Fernandes, J.G.; Rodnight, R. Wernicke-Korsakoff syndrome. Postgrad. Med. J. 1997, 73, 27–31.
- Gibson, G.E.; Hirsch, J.A.; Fonzetti, P.; Jordan, B.D.; Cirio, R.T.; Elder, J. Vitamin B1 (thiamine) and dementia. Ann. N. Y. Acad. Sci. 2016, 1367, 21–30.
- Gold, M.; Hauser, R.A.; Chen, M.F. Plasma thiamine deficiency associated with Alzheimer’s disease but not Parkinson’s disease. Metab. Brain Dis. 1998, 13, 43–53.
- Wilkinson, T.J.; Hanger, H.C.; George, P.M.; Sainsbury, R. Is thiamine deficiency in elderly people related to age or co-morbidity? Age Ageing 2000, 29, 111–116.
- Lazarov, J. Resorption of vitamin B1—XII. Changes in the resorption and the phosphorylation of thiamine in rats in relation to age. Exp. Gerontol. 1977, 12, 75–79.
- Krebs, H.A. The regulation of the release of ketone bodies by the liver. Adv. Enzym. Regul. 1966, 4, 339–354.
- Walsh, E.I.; Shaw, M.; Sachdev, P.; Anstey, K.J.; Cherbuin, N. Brain atrophy in ageing: Estimating effects of blood glucose levels vs. other type 2 diabetes effects. Diabetes Metab. 2018, 44, 80–83.
- Nedelska, Z.; Schwarz, C.G.; Boeve, B.F.; Lowe, V.J.; Reid, R.I.; Przybelski, S.A.; Lesnick, T.G.; Gunter, J.L.; Senjem, M.L.; Ferman, T.J.; et al. White matter integrity in dementia with Lewy bodies: A voxel-based analysis of diffusion tensor imaging. Neurobiol. Aging 2015, 36, 2010–2017.
- Ivanov, A.I.; Malkov, A.E.; Waseem, T.; Mukhtarov, M.; Buldakova, S.; Gubkina, O.; Zilberter, M.; Zilberter, Y. Glycolysis and oxidative phosphorylation in neurons and astrocytes during network activity in hippocampal slices. J. Cereb. Blood Flow. Metab. 2014, 34, 397–407.
- Pellerin, L.; Magistretti, P.J. Neuroscience. Let there be (NADH) light. Science 2004, 305, 50–52.
- Demetrius, L.A.; Simon, D.K. An inverse-Warburg effect and the origin of Alzheimer’s disease. Biogerontology 2012, 13, 583–594.
- Demetrius, L.A.; Magistretti, P.J.; Pellerin, L. Alzheimer’s disease: The amyloid hypothesis and the Inverse Warburg effect. Front. Physiol. 2014, 5, 522.
- Chandrasekaran, K.; Hatanpaa, K.; Brady, D.R.; Rapoport, S.I. Evidence for physiological down-regulation of brain oxidative phosphorylation in Alzheimer’s disease. Exp. Neurol. 1996, 142, 80–88.
- Lehmann, M.; Ghosh, P.M.; Madison, C.; Karydas, A.; Coppola, G.; O’Neil, J.P.; Huang, Y.; Miller, B.L.; Jagust, W.J.; Rabinovici, G.D. Greater medial temporal hypometabolism and lower cortical amyloid burden in ApoE4-positive AD patients. J. Neurol. Neurosurg. Psychiatry 2014, 85, 266–273.
- Wu, H.; Zhou, S.; Zhao, H.; Wang, Y.; Chen, X.; Sun, X. Effects of apolipoprotein E gene polymorphism on the intracellular Ca2+ concentration of astrocytes in the early stages post injury. Exp. Med. 2018, 15, 1417–1423.
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 14.
- Nagy, Z.; Esiri, M.M.; Jobst, K.A.; Johnston, C.; Litchfield, S.; Sim, E.; Smith, A.D. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease. Neuroscience 1995, 69, 757–761.
- Butterfield, D.A.; Lange, M.L. Multifunctional roles of enolase in Alzheimer’s disease brain: Beyond altered glucose metabolism. J. Neurochem. 2009, 111, 915–933.
- Castegna, A.; Aksenov, M.; Aksenova, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: Creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic. Biol. Med. 2002, 33, 562–571.
- Castegna, A.; Aksenov, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: Dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J. Neurochem. 2002, 82, 1524–1532.
- Inoue, Y.; Tasaki, M.; Masuda, T.; Misumi, Y.; Nomura, T.; Ando, Y.; Ueda, M. alpha-Enolase reduces cerebrovascular Abeta deposits by protecting Abeta amyloid formation. Cell. Mol. Life Sci. 2022, 79, 462.
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20.
- Vanitallie, T.B. Preclinical sporadic Alzheimer’s disease: Target for personalized diagnosis and preventive intervention. Metabolism 2013, 62 (Suppl. S1), S30–S33.
- Mosconi, L.; Brys, M.; Switalski, R.; Mistur, R.; Glodzik, L.; Pirraglia, E.; Tsui, W.; De Santi, S.; de Leon, M.J. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc. Natl. Acad. Sci. USA 2007, 104, 19067–19072.
- Mosconi, L.; Tsui, W.H.; De Santi, S.; Li, J.; Rusinek, H.; Convit, A.; Li, Y.; Boppana, M.; de Leon, M.J. Reduced hippocampal metabolism in MCI and AD: Automated FDG-PET image analysis. Neurology 2005, 64, 1860–1867.
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530.
- Mullins, R.J.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 118.
- Ogata, S.; Ito, S.; Masuda, T.; Ohtsuki, S. Changes of Blood-Brain Barrier and Brain Parenchymal Protein Expression Levels of Mice under Different Insulin-Resistance Conditions Induced by High-Fat Diet. Pharm. Res. 2019, 36, 141.
- Soto, M.; Cai, W.; Konishi, M.; Kahn, C.R. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc. Natl. Acad. Sci. USA 2019, 116, 6379–6384.
- Sathya, M.; Premkumar, P.; Karthick, C.; Moorthi, P.; Jayachandran, K.S.; Anusuyadevi, M. BACE1 in Alzheimer’s disease. Clin. Chim. Acta 2012, 414, 171–178.
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43.
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin resistance and pathological brain ageing. Diabet. Med. 2011, 28, 1463–1475.
- Cai, H.; Cong, W.N.; Ji, S.; Rothman, S.; Maudsley, S.; Martin, B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr. Alzheimer Res. 2012, 9, 5–17.
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595.
- Frankiewicz, T.; Parsons, C.G. Memantine restores long term potentiation impaired by tonic N-methyl-D-aspartate (NMDA) receptor activation following reduction of Mg2+ in hippocampal slices. Neuropharmacology 1999, 38, 1253–1259.
- Tominaga-Yoshino, K.; Uetsuki, T.; Yoshikawa, K.; Ogura, A. Neurotoxic and neuroprotective effects of glutamate are enhanced by introduction of amyloid precursor protein cDNA. Brain Res. 2001, 918, 121–130.
- Braak, H.; Braak, E.; Ohm, T.; Bohl, J. Alzheimer’s disease: Mismatch between amyloid plaques and neuritic plaques. Neurosci. Lett. 1989, 103, 24–28.
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J. Neurochem. 2009, 111, 242–249.
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481.
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364.
- Medina, D.; DeToledo-Morrell, L.; Urresta, F.; Gabrieli, J.D.; Moseley, M.; Fleischman, D.; Bennett, D.A.; Leurgans, S.; Turner, D.A.; Stebbins, G.T. White matter changes in mild cognitive impairment and AD: A diffusion tensor imaging study. Neurobiol. Aging 2006, 27, 663–672.
- Kuczynski, B.; Targan, E.; Madison, C.; Weiner, M.; Zhang, Y.; Reed, B.; Chui, H.C.; Jagust, W. White matter integrity and cortical metabolic associations in aging and dementia. Alzheimers Dement. 2010, 6, 54–62.
- O’Dwyer, L.; Lamberton, F.; Bokde, A.L.; Ewers, M.; Faluyi, Y.O.; Tanner, C.; Mazoyer, B.; O’Neill, D.; Bartley, M.; Collins, D.R.; et al. Multiple indices of diffusion identifies white matter damage in mild cognitive impairment and Alzheimer’s disease. PLoS ONE 2011, 6, e21745.
- Goldman-Rakic, P.S.; Selemon, L.D.; Schwartz, M.L. Dual pathways connecting the dorsolateral prefrontal cortex with the hippocampal formation and parahippocampal cortex in the rhesus monkey. Neuroscience 1984, 12, 719–743.
- Risacher, S.L.; Saykin, A.J.; West, J.D.; Shen, L.; Firpi, H.A.; McDonald, B.C.; Alzheimer’s Disease Neuroimaging, I. Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Curr. Alzheimer Res. 2009, 6, 347–361.
- Villain, N.; Fouquet, M.; Baron, J.C.; Mezenge, F.; Landeau, B.; de La Sayette, V.; Viader, F.; Eustache, F.; Desgranges, B.; Chetelat, G. Sequential relationships between grey matter and white matter atrophy and brain metabolic abnormalities in early Alzheimer’s disease. Brain 2010, 133, 3301–3314.
- Sokoloff, L. Metabolism of ketone bodies by the brain. Annu. Rev. Med. 1973, 24, 271–280.
- Young, K.J.; Bennett, J.P. The mitochondrial secret(ase) of Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S381–S400.
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21–35.
- Fields, R.D. White matter in learning, cognition and psychiatric disorders. Trends. Neurosci. 2008, 31, 361–370.
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449.
- Calingasan, N.Y.; Gibson, G.E. Vascular endothelium is a site of free radical production and inflammation in areas of neuronal loss in thiamine-deficient brain. Ann. N. Y. Acad. Sci. 2000, 903, 353–356.
- Hazell, A.S.; Butterworth, R.F. Update of cell damage mechanisms in thiamine deficiency: Focus on oxidative stress, excitotoxicity and inflammation. Alcohol. Alcohol. 2009, 44, 141–147.
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770.
- Moley, K.H.; Mueckler, M.M. Glucose transport and apoptosis. Apoptosis 2000, 5, 99–105.
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Soininen, H.; Hiltunen, M. AMP-activated protein kinase: A potential player in Alzheimer’s disease. J. Neurochem. 2011, 118, 460–474.
- Dice, J.F. Chaperone-mediated autophagy. Autophagy 2007, 3, 295–299.
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199.
- Lafay-Chebassier, C.; Paccalin, M.; Page, G.; Barc-Pain, S.; Perault-Pochat, M.C.; Gil, R.; Pradier, L.; Hugon, J. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem. 2005, 94, 215–225.
- Bove, J.; Martinez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 2011, 12, 437–452.
- Hamano, T.; Gendron, T.F.; Causevic, E.; Yen, S.H.; Lin, W.L.; Isidoro, C.; Deture, M.; Ko, L.W. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur. J. Neurosci. 2008, 27, 1119–1130.
- Freude, S.; Schilbach, K.; Schubert, M. The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer’s disease: From model organisms to human disease. Curr. Alzheimer Res. 2009, 6, 213–223.
- Schwartz, M.W.; Figlewicz, D.F.; Kahn, S.E.; Baskin, D.G.; Greenwood, M.R.; Porte, D., Jr. Insulin binding to brain capillaries is reduced in genetically obese, hyperinsulinemic Zucker rats. Peptides 1990, 11, 467–472.
- Hoyer, S. The aging brain. Changes in the neuronal insulin/insulin receptor signal transduction cascade trigger late-onset sporadic Alzheimer disease (SAD). A mini-review. J. Neural. Transm. 2002, 109, 991–1002.
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80.
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338.
- Subramanian, S.; John, M. Intranasal administration of insulin lowers amyloid-beta levels in rat model of diabetes. Indian J. Exp. Biol. 2012, 50, 41–44.
- Van den Berg, E.; Kloppenborg, R.P.; Kessels, R.P.; Kappelle, L.J.; Biessels, G.J. Type 2 diabetes mellitus, hypertension, dyslipidemia and obesity: A systematic comparison of their impact on cognition. Biochim. Biophys. Acta 2009, 1792, 470–481.
- Bruning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Muller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125.
- Fisher, S.J.; Bruning, J.C.; Lannon, S.; Kahn, C.R. Insulin signaling in the central nervous system is critical for the normal sympathoadrenal response to hypoglycemia. Diabetes 2005, 54, 1447–1451.
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468.
- Kappeler, L.; De Magalhaes Filho, C.; Dupont, J.; Leneuve, P.; Cervera, P.; Perin, L.; Loudes, C.; Blaise, A.; Klein, R.; Epelbaum, J.; et al. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 2008, 6, e254.
- Heidenreich, K.A.; Toledo, S.P.; Kenner, K.A. Regulation of protein phosphorylation by insulin and insulin-like growth factors in cultured fetal neurons. Adv. Exp. Med. Biol. 1991, 293, 379–384.
- Lee, C.C.; Huang, C.C.; Wu, M.Y.; Hsu, K.S. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J. Biol. Chem. 2005, 280, 18543–18550.
- Bramham, C.R.; Messaoudi, E. BDNF function in adult synaptic plasticity: The synaptic consolidation hypothesis. Prog. Neurobiol. 2005, 76, 99–125.
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365.
- Prakash, A.; Kumar, A. Role of nuclear receptor on regulation of BDNF and neuroinflammation in hippocampus of beta-amyloid animal model of Alzheimer’s disease. Neurotox. Res. 2014, 25, 335–347.
- Schur, E.A.; Melhorn, S.J.; Oh, S.K.; Lacy, J.M.; Berkseth, K.E.; Guyenet, S.J.; Sonnen, J.A.; Tyagi, V.; Rosalynn, M.; De Leon, B.; et al. Radiologic evidence that hypothalamic gliosis is associated with obesity and insulin resistance in humans. Obesity 2015, 23, 2142–2148.
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 2015, 64, 3927–3936.
- Draznin, B. Molecular mechanisms of insulin resistance: Serine phosphorylation of insulin receptor substrate-1 and increased expression of p85alpha: The two sides of a coin. Diabetes 2006, 55, 2392–2397.
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 2010, 93, 546–553.
- Sivanesan, S.; Mundugaru, R.; Rajadas, J. Possible Clues for Brain Energy Translation via Endolysosomal Trafficking of APP-CTFs in Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2018, 2018, 2764831.
- Velliquette, R.A.; O’Connor, T.; Vassar, R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: Possible early events in Alzheimer’s disease pathogenesis. J. Neurosci. 2005, 25, 10874–10883.
- Moreira, P.I.; Cardoso, S.M.; Pereira, C.M.; Santos, M.S.; Oliveira, C.R. Mitochondria as a therapeutic target in Alzheimer’s disease and diabetes. CNS Neurol. Disord. Drug Targets 2009, 8, 492–511.
- Giese, K.P. GSK-3: A key player in neurodegeneration and memory. IUBMB Life 2009, 61, 516–521.
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439.
- Alvarez, G.; Munoz-Montano, J.R.; Satrustegui, J.; Avila, J.; Bogonez, E.; Diaz-Nido, J. Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett. 1999, 453, 260–264.
- Hooper, C.; Markevich, V.; Plattner, F.; Killick, R.; Schofield, E.; Engel, T.; Hernandez, F.; Anderton, B.; Rosenblum, K.; Bliss, T.; et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 2007, 25, 81–86.
- Hoshi, M.; Takashima, A.; Noguchi, K.; Murayama, M.; Sato, M.; Kondo, S.; Saitoh, Y.; Ishiguro, K.; Hoshino, T.; Imahori, K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc. Natl. Acad. Sci. USA 1996, 93, 2719–2723.
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci 2001, 21, 2561–2570.
- Townsend, M.; Mehta, T.; Selkoe, D.J. Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J. Biol. Chem. 2007, 282, 33305–33312.
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976.
- Lee, C.C.; Kuo, Y.M.; Huang, C.C.; Hsu, K.S. Insulin rescues amyloid beta-induced impairment of hippocampal long-term potentiation. Neurobiol. Aging 2009, 30, 377–387.
- Zhao, L.; Teter, B.; Morihara, T.; Lim, G.P.; Ambegaokar, S.S.; Ubeda, O.J.; Frautschy, S.A.; Cole, G.M. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: Implications for Alzheimer’s disease intervention. J. Neurosci. 2004, 24, 11120–11126.
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62.
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21.
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969.
- Marciniak, E.; Leboucher, A.; Caron, E.; Ahmed, T.; Tailleux, A.; Dumont, J.; Issad, T.; Gerhardt, E.; Pagesy, P.; Vileno, M.; et al. Tau deletion promotes brain insulin resistance. J. Exp. Med. 2017, 214, 2257–2269.
- Kerr, F.; Rickle, A.; Nayeem, N.; Brandner, S.; Cowburn, R.F.; Lovestone, S. PTEN, a negative regulator of PI3 kinase signalling, alters tau phosphorylation in cells by mechanisms independent of GSK-3. FEBS Lett. 2006, 580, 3121–3128.
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e5.

