1. Insulin Resistance Is Linked to AD
AD is so closely related to diabetes that it is known as a so-called ‘type 3 diabetes’. Decrease in brain glucose metabolism occurs more than 10 years before the onset of AD symptoms [
12]. Therefore, abnormal energy metabolism is pointed out as the cause of dementia. According to the results of a Whitehall II cohort study in the UK (n = 5653), memory decreased 45% faster, rational judgment ability declined 29% faster, and global cognitive ability to evaluate multiple cognitive functions using ACE-R score decreased 24% more rapidly in patients with diabetes [
110]. In middle-aged populations with type 2 diabetes, the duration of illness and the degree of glycemic dysregulation had a significant effect on the rate of cognitive decline [
110].
1.1. Brain Energy Mobilization
Among the organs of the body, the brain consumes one of the largest proportions of energy. Although brain mass constitutes 2% of total body weight, its glucose consumption in the resting awake state accounts for 25% of total body glucose used [
111]. A significant amount of this energy is used to maintain the nerve cell membrane potential which is important for signal conduction [
112]. The brain uses glucose, lactate, ketone bodies, amino acids, and short chain fatty acids (SCFAs) as energy sources, but glucose is used preferentially, and the others are utilized as alternative energy sources in the absence of glucose [
113].
The brain uses 6–7 mg/100 g of glucose per minute, which is equivalent to 120–130 g per day [
114]. Because glucose is a polar substance, it cannot freely pass through the cell membrane but enters the cell through a transporter called glucose transporter (GLUT). In the brain, GLUT1 and GLUT3 are the main glucose transporters, and are not insulin dependent type transporters [
115]. Insulin is a pancreatic hormone that lowers blood glucose by acting on GLUT4 to transport glucose into cells. Insulin resistance is a phenomenon in which this action of insulin is weakened and blood glucose concentration increases [
116]. In the brain, GLUT4 is far less prevalent than GLUT1 and GLUT3, which means that the brain is not an organ that stores glucose in response to insulin. However, it is known that GLUT4 is involved in glucose influx into synaptic areas during high synaptic activity conditions [
117]. Therefore, although insulin resistance does not reduce the overall influx of glucose into brain tissue, it may affect synaptic activity.
Lactate is the product of anaerobic glycolysis and is mainly produced in skeletal muscles that require a rapid supply of ATP [
118]. Lactic acid produced in the muscles enters systemic circulation and goes to the liver, where it is reconverted into glucose for recycling. Circulating lactic acid can also enter brain cells through the MCT (monocarboxylate transporter) channel to be used as an energy source [
119]. Lactic acid is also produced by astrocytes during glucose metabolism to provide ATP for the conversion of glutamate into glutamine [
120]. Lactic acid produced in astrocytes goes to nerve cells and is converted into pyruvate. Astrocytes play an important role in creating emotionally salient memory by synthesizing glycogen using glucose and supplying it to neurons by making lactate [
121].
Ketone bodies are the general name of acetone, acetoacetate, and β-hydroxybutyrate. When blood sugar decreases, such as in continuous fasting, the liver breaks down triglycerides and produces free fatty acid and ketone bodies to provide an alternative energy source [
122]. However, due to their long chain structure, fatty acids hardly pass the BBB, whereas ketones have a simple structure and can pass through the BBB. These ketone bodies go to nerve cells and are converted into acetyl-CoA and utilized for energy production [
123].
Amino acid is the main component of the structure and function of a living body rather than an energy source. However, in a low energy state, by removing the amine group, an amino acid can be converted to acetyl-CoA or other intermediate metabolite of the TCA cycle. This process is called gluconeogenesis [
124].
SCFA is a substance produced by decomposition of dietary fiber in the digestive tract by enterobacteriaceae, and three main types have been identified: acetate, propionate, and butyrate [
125]. They are absorbed by the intestinal epithelial cells and utilized as energy sources. Butyrate is mostly consumed by enterocytes (gastrointestinal epithelial cells), propionate can reach to the liver and is generally utilized for gluconeogenesis by hepatocytes, and acetate reaches the blood circulation. Of these, butyrate has many known functions in the brain, but because it hardly reaches the circulation, the amount that passes through the BBB is little [
126]. However, the amount of butyrate in the blood may increase depending on the intake of dietary fiber or the characteristics of enterobacteriaceae [
127]. Currently, research on the effects of SCFAs and gut-brain interaction is being actively conducted.
For the delivery of these energy sources, passage is required through the neurovascular unit composed of capillaries and cells in the brain. This unit is composed of vascular endothelial cells, pericytes, basal lamina, astrocytes, and neurons [
128]. The BBB, which consists of the tight junction of vascular endothelial cells, basement membrane, and end foot of astrocytes, can pass lipid-soluble substances, but water-soluble substances such as glucose, amino acids, and nucleic acids need transport proteins to transit through [
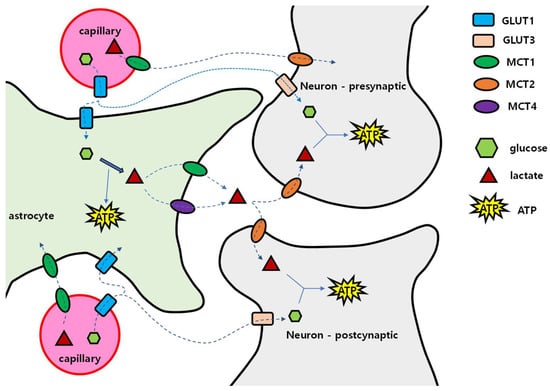
129]. Once glucose has crossed the BBB, glucose can reach neurons through two pathways: via the extracellular fluid in brain tissue, and via astrocytes (
Figure 1). In the extracellular pathway, glucose enters GLUT1 of the vascular endothelial cells, spreads into the extracellular fluid, and then enters neurons through GLUT3. In the astrocytic pathway, after passing through GLUT1 of vascular endothelial cells, glucose enters astrocytes through GLUT1, and is converted to glycogen for storage or to lactic acid by glycolysis. Then, lactic acid exits into extracellular fluid by MCT1 or MCT4. In the extracellular fluid, lactic acid enters the nerve cell through MCT2. This is called the ‘astrocyte–neuron lactate shuttle’.
Figure 1. Glucose and lactate delivery to nerve cells. Glucose is transported from capillaries to astrocytes via GLUT1. After entering the astrocyte, glucose is metabolized to lactate, then lactate is transported out into the interstitium via MCT1 and MCT4. Lactate enters into presynaptic and postsynaptic neurons via MCT2 and is used for energy metabolism after being converted into pyruvate. Glucose can also enter neurons directly via GLUT3. Blood capillary lactate enters into astrocytes via MCT1 and into neurons via MCT2. GLUT: glucose transporters; MCT: monocarboxylate transporter.
Concentrations of glucose and lactic acid in the brain extracellular fluid are much lower than in blood, which means that concentration gradient may drive the diffusion of these molecules [
130]. Then, if the concentration of glucose and lactic acid in the blood increases, the concentration of glucose and lactic acid in the cerebrospinal fluid will also increase. However, in patients with type 2 diabetes, the glucose concentration in the cerebrospinal fluid was lower than in normoglycemic persons, which may be because systemic insulin resistance induced intracerebral insulin resistance [
131]. Additionally, in the glycolysis process of glucose, enzyme activity is regulated through several steps. Hexokinase initiates the glycolytic process by converting glucose into glucose-6-phosphate, and when this enzyme is saturated (Km = 0.05 mmol/L), glycolysis no longer occurs even when the amount of glucose increases. On the other hand, the reaction of lactate conversion into pyruvate only requires NAD
+ and can be used for ATP synthesis [
132]. Unlike glucose, lactic acid has no restriction on conversion to pyruvate, so it is useful as a continuous energy source for nerve cells.
Astrocytes convert glutamate into glutamine [
133]. The energy required for this reaction comes from the metabolism of glucose to lactate. Therefore, when the glucose supply to astrocytes is reduced, excitotoxicity may occur due to poor glutamate removal from excitatory synapses. Memantine, an NMDA antagonist approved for AD treatment, may suppress excitotoxicity in a status of excessive synaptic glutamate which may be a consequence of insufficient glucose [
134]. Generally, among the cells in the brain, only astrocytes synthesize and store glycogen, and energy production by gluconeogenesis which uses aspartate, glutamate, alanine, and lactate only occurs in astrocytes but not in neurons [
135]. Nerve cells are vulnerable to a low energy supply and therefore depend on other cells in the brain tissue.
1.2. When and How the Brain Deals with the Lack of Glucose
In AD, hypometabolic changes precede the appearance of dementia symptoms by a decade or more. There are other conditions associated with brain hypometabolism such as Beriberi. Beriberi is a hypometabolic state due to the lack of thiamine (vitamin B1) [
136]. Thiamine is a material used for the synthesis of thiamine diphosphate (TPP), and TPP is required for the enzymatic activity of pyruvate dehydrogenase complex and the ketoglutarate dehydrogenase complex. These two enzyme complexes play a key role in oxidative phosphorylation. The pyruvate dehydrogenase converts pyruvate to acetyl-CoA, and the ketoglutarate dehydrogenase converts α-ketoglutarate to succinyl-CoA; these two CoAs are the intermediate molecules of the TCA cycle. Therefore, when thiamine is insufficient, these enzymes do not work, and the cellular energy factory producing ATPs shuts down. Another enzyme that uses TPP as a coenzyme is transketolase, which mediates the non-oxidative pentose phosphate pathway (PPP) [
137]. The PPP makes hexose, a constituent of DNA and RNA, from glucose. That is to say, thiamine deficiency causes a shortage of materials for DNA and RNA synthesis.
The causative factors for Beriberi include long-term malnutrition due to alcoholism, malabsorption of thiamine, pregnancy, increased thiamine requirement due to hyperthyroidism, decreased thiamine absorption due to liver disease, and loss of thiamine due to dialysis or chronic diarrhea [
138]. Thiamine deficiency causes memory impairment, amyloidosis and hyperphosphorylation of tau in the brain [
84,
139]. Severe Beriberi, called Wernicke–Korsakoff syndrome, results in permanent damage of the central nervous system, and mainly occurs in chronic alcohol abuse patients [
140]. In animal studies, thiamine deficiency induced dementia-like conditions such as decreased neurogenesis, memory loss, and plaque/tangle formation [
141]. In AD patients, the amount of thiamine was lowered by one-third compared to age matched normal controls [
142]. Additionally, the concentration of thiamine gradually decreases with age [
143]. The cause of the decrease in thiamine concentration with age is not yet clear, but it was suggested that a gradual decrease in intestinal alkaline phosphatase activity may lead to diminished thiamine absorption [
144].
When the amount of glucose in the bloodstream decreases, the liver breaks down stored fat to make ketone bodies, which become the main energy source for the brain [
145]. However, if the supply of ketones is inadequate, brain cells break down the myelin sheath, which contains a lot of lipids, to produce ketones on their own. This process may lead to the decrease in white matter volume that occurs with age [
146]. Additionally, the decrease in white matter volume becomes more pronounced as dementia progresses, suggesting that dementia is associated with a lack of energy in the brain [
147].
Changes in energy metabolism in the brain also occur during the normal aging process. In the brain, ATP is synthesized in astrocytes and neurons, with astrocytes primarily using the glycolysis pathway and neurons producing ATP through oxidative phosphorylation [
148]. However, the brain under cellular stress requires more energy, and to supplement the insufficient ATP, the astrocytes increase glycolysis to produce and secrete more lactate into the brain interstitium [
149]. Neurons absorb and convert this lactate into acetyl-CoA to make ATP through oxidative phosphorylation. Among these neurons, some maintain a normal state, but others with oxidative damage have a compensatory increase in oxidative phosphorylation, and were described by Demetrius et al., as type 2 neurons [
150]. When the supply of lactate is increased, these type 2 neurons synthesize more ATP, which is called the ‘inverse-Warberg effect’ and appears in aged neurons [
151]. However, due to mitochondrial damage, more oxygen free radicals are made and more oxidative damage to the surrounding cells occurs [
150]. When such damage accumulates, neuronal functions deteriorate as evidenced by decreased ATP production and the pathological process progresses as the function of the cells gradually decreases. Sporadic AD is also suggested as a disease related to insufficient energy metabolism [
152].
Cortical hypometabolism also appears before the onset of AD in the ApoE4 carrier, which has been pointed out as a genetic factor associated with the onset of AD [
153]. Among the three types of human apolipoprotein Es, ApoE4 showed the lowest intracerebral glucose uptake and metabolism [
154]. Additionally, the upregulation of oxidative phosphorylation in neurons happens more in the ApoE4 carriers [
155]. On the other hand, intracerebral movement of ketone bodies was the least with ApoE3. This may be related to the reason why ApoE2 has the lowest incidence of AD, and ApoE4 carriers have increased AD incidence [
156].
Enolase, an enzyme whose activity was changed in MCI and AD, is involved in glucose metabolism [
157]. Enolase converts 2-phosphoglycerate to phosphoenolpyruvate in the glycolysis pathway. Interestingly, enolase activity was increased in MCI, early onset AD, and AD [
158,
159]. The increase in enolase activity may be an adaptive process in which glucose is more efficiently decomposed and supplied as an energy source in a state of insufficient glucose. In addition, enolase increases tPA binding and increases plasmin, and this can degrade Aβ [
160]. Therefore, the increase in enolase activity may be a sign of abnormal brain metabolism and can be seen as an effort to eliminate Aβ.
2. How Insulin Resistance Causes Dementia
Due to insulin resistance, AD-related pathological process progresses due to (1) the amount of glucose is insufficient in the brain and (2) the intracellular insulin signaling is weakened.
2.1. Brain Glucose Insufficiency
The storage capacity of glucose in the brain is limited, so when the glucose supply is reduced, brain function deteriorates rapidly [
238]. Vanitallie (2013) showed that hypometabolic state of brain glucose precedes cognitive symptoms in dementia [
239]. Glucose consumption reduces first in the hippocampus, followed by the posterior cingulate cortex, parietal lobe, and frontal lobe [
240]. Additionally, there is an apparent decrease in glucose metabolism in hippocampal structures based on FDG-PET scan images in MCI patients [
241]. In normal aged individuals, sometimes glucose metabolism is decreased but not always, which leads to the speculation that the decreased glucose metabolism in MCI is likely pathologic rather than an aging process [
238]. Furthermore, it was found that the decrease in glucose metabolism in AD patients was proportional to the decrease in GLUT1 expression [
242]. As a mechanism for this, insulin resistance due to high-glycemic diet decreased brain GLUT1 expression and induced AD-like symptoms [
243,
244]. Glucose intolerance resulting from inactivation of both insulin receptor and IGF-1 receptors in the hippocampus or central amygdala caused cognitive deficits and anxiety [
245].
When energy metabolism in the brain decreases, BACE1 activity increases and results in more Aβ accumulates [
246]. Similar lack of energy metabolism is seen in thiamine deficiency, which causes an increase in BACE1 activity and Aβ accumulation [
141]. Decreased activity in the electron transport chain due to the lack of intracerebral glucose also leads to increased production of APP and accumulation of Aβ [
247]. ROS overproduction due to mitochondrial dysfunction enhances the accumulation of Aβ and induces oxidative damage to proteins, lipids, and nucleic acids [
1,
247].
Impaired glucose metabolism is also associated with excitotoxicity as a result of reduced glutamate uptake in astrocytes [
248,
249]. As NMDA receptor antagonist memantine has been shown to improve AD patients’ cognitive functions, excitotoxicity may have a role in AD pathology [
250]. It was also shown that Aβ oligomers interact with NMDA receptor to exert neurotoxicity [
251]. In post-mortem AD brains, amyloid plaques, NFTs are co-localized with excitatory pyramidal neurons which support the notion that excitotoxicity is involved in the pathological mechanism of AD [
252].
When the amount of intracellular glucose decreases by reduced GLUT1 and GLUT3 expression, the amount of uridine 5’-diphosphate-N-acetylglucosamine (UDP-GlcNAc), which is a product of glucose metabolism, is reduced [
253]. UDP-GlcNAc binds to tau and APP by O-GlcNAc transferase (OGT), and O-GlcNAcylation prevents phosphorylation of these proteins. Therefore, when the amount of UDP-GlcNAc is decreased, hyperphosphorylation of tau and APP can occur, resulting in neurotoxicity [
254]. Because the reciprocal changes in O-GlcNAylation and hyperphsohorylation of tau responds rapidly to glucose availability, decreasing brain glucose uptake could contribute to the development of AD [
255].
Decreased white matter density is another early sign of AD and MCI [
256]. Changes in the intensity of white matter are also related to energy metabolism [
257]. The areas where these changes occur mainly are the cingulum bundle, uncinate fasciculus, and superior longitudinal fasciculus of MCI patients, which are the areas corresponding to the DMN [
258]. In particular, the cingulum bundle connects the hippocampal formation, prefrontal cortex, and posterior cingulate cortex [
259]. Patients with MCI also have hypometabolism in the PFC and posterior cingulate cortex, which are also the main components of DMN [
258]. Connectivity between these areas is provided by the superior longitudinal fasciculus [
257]. Therefore, degeneration of the cingulum and hypometabolism of the prefrontal cortex are associated with hippocampus atrophy and cognitive decline [
260,
261]. When glucose is insufficient, ketone bodies are used as an alternative brain energy source [
262]. This loss in white matter integrity could be a direct result of a switch from the use of ketone body supplied from the peripheral ketogenic organ, the liver, to ketone body resulting from local myelin breakdown via FA oxidation by astroglia [
122]. In the prodromal AD brain, ketogenic enzyme such as SCOT (3-ketoacid CoA transferase) is upregulated [
263]. Since insulin suppresses ketone body production in the liver, in a systemic insulin resistant state, ketone supplement to the brain is not enough. Then, the brain cells try to obtain an energy source through catabolic gluconeogenesis [
264]. Decreased mitochondrial efficiency, increased oxidative stress and H
2O
2 overproduction activate PLA2 (phospholipase 2) and result in myelin sheath degradation for use of fatty acid and ketogenesis [
122]. If this process continues for a long time, the myelin sheath of the white matter decreases, and the speed of nerve transmission through action potentials between neurons also decreases [
265]. Free radical induced mitochondrial damage and accumulation of Aβ accelerate this process of energy anomaly as a vicious cycle [
266]. Alternately, lesions in white matter integrity may be caused by inadequate lipid synthesis due to competition between consumption of ketones/acetyl-CoA for bioenergetics and lipid synthesis [
122].
Impaired glucose metabolism could induce inflammatory responses and exacerbate AD pathology. Pathophysiological cascades of inflammatory responses are associated with mitochondrial dysfunction and oxidative stress, excess of inflammatory factors, excitotoxicity, AGEs, apoptosis, hyper-activation of protein kinases, etc. [
84,
267,
268,
269,
270,
271]. Additionally, glucose metabolism is necessary for autophagy. Autophagy is responsible for the degradation of folded proteins in cells, and its dysfunction could lead to Aβ aggregation and tauopathy [
272]. The initiator of autophagy process, Beclin 1, was decreased in AD patients [
273]. The mammalian target of rapamycin (mTOR) pathway receives autophagic stimuli and signals to initiate autophagy [
248]. Enhanced mTOR signaling activity increased Aβ deposits and NFT formation while inhibition of mTOR by rapamycin reduced Aβ pathology by increasing autophagy [
274,
275]. Impairment of autophagy also increased β- and γ-secretase activities and contributes to the tauopathy [
248,
276].
2.2. Brain Insulin Insufficiency
As the blood insulin concentration increases, the brain and CSF insulin concentrations also increase correspondingly, but in a prolonged hyperinsulinemia state, insulin receptor of the BBB downregulates and insulin transport into the brain reduces [
277]. Insulin receptors are distributed in astrocytes and neuronal synapses, especially in the olfactory bulb, cingulate cortex, hippocampus, hypothalamus, amygdala and septum [
278].
Based on the results from the previous studies, insulin receptors, insulin-like growth factor receptors, and IRS-1 participates in the pathogenesis of AD [
279]. Steen et al., (2005) has demonstrated that the expression of insulin and IGF-1/2 receptors were markedly reduced in AD brains in correlation with the pathological alterations of AD such as increased GSK-3β activity and APP mRNA expression [
280]. Moreover, IRS-1 was suppressed in the hippocampus, and the degree of suppression was proportional to the amount of senile plaque and cognitive decline [
281]. Accordingly, intranasal insulin injection improved cognitive function in AD patients [
282].
As the brain is an insulin responding organ, insulin resistance correlates to cognitive dysfunction [
283]. When the brain has a decreased number of insulin receptors, hyperphagia, insulin resistance, central hypogonadism, impaired response to hypoglycemia, and depression-like behaviors appear whereas loss of IGF-1 receptor caused impaired brain development [
284,
285,
286,
287]. Insulin resistance also results in the lack of trophic support. In the brain, insulin/p-IRS/PI3K/Akt signaling enhances neurite outgrowth and synaptogenesis via upregulating BDNF and PSD-95 [
288,
289]. BDNF promotes synaptic plasticity through CaMKII, synaptophysin and PSD-95 [
290]. In an insulin resistant condition, activation of microglia and astrocytes continues, and these cells secrete glia-derived proinflammatory cytokine which lowers BDNF levels [
291,
292,
293]. Insulin resistance model animals showed impaired hippocampal neuroplasticity which is characterized by an increase in PI3K p85 subunit autophosphorylation with a decrease in phospho-Akt [
194,
294]. When insulin receptors and IGF-1 receptors were inactivated in the hippocampus (Hippo-DKO), GluR1 expression decreased and increased anxiety, cognitive impairment, and systemic glucose intolerance were shown [
245]. These finding demonstrate that the brain requires insulin signaling to maintain its regular functions and the impact of insulin resistance is not only a systemic effect but also directly affects the brain [
295].
In a hypometabolic state, amyloidosis increases based on the findings in animal studies and in vitro experiments [
296]. Low glucose metabolism and insulin resistance in the brain stimulated BACE1 and GSK-3 activity [
297,
298]. GSK-3β activation increased APP mRNA expression, BACE activity, tau phosphorylation, Aβ aggregation, and memory impairment, as well as microglia activation-associated inflammatory reactions in AD [
298,
299]. GSK-3α can modulate APP cleavage and induce Aβ production and that blockade of GSK-3β could prevent Aβ accumulation [
300,
301]. Overexpression of GSK-3 suppressed LTP by negatively regulating Wnt or PI3K signaling which can lead to memory impairments [
302]. GSK-3β also reduced acetylcholine synthesis and induced apoptosis of cholinergic neurons resulting in NFT formation [
303].
Insulin accelerates the movement of Aβ from the Golgi network to the plasma membrane, thereby helping Aβ to be released out of neuronal cells [
304]. Conversely, Aβ binds to the insulin receptor and inhibits insulin signaling, resulting in LTP inhibition and neuronal spine loss [
305]. As a result, insulin suppresses the toxicity caused by Aβ binding to the synaptic insulin receptor [
306]. Insulin can also suppress the formation of Aβ oligomer [
307]. In addition, insulin has been proposed to regulate extracellular degradation of Aβ by modulating the IDE activity [
308]. If the amount of brain insulin decreases, IDE activity also decreases resulting in an increase of Aβ [
308]. Additionally, when PI3K/AKT activity is decreased due to decreased insulin signaling, o-glcNAcylation is also decreased, resulting in hyperphosphorylation of tau [
309].
Tauopathy is causes cognitive dysfunction by synaptic plasticity disorders and degenerative lesions [
310]. In AD tauopathy develops in the brainstem and entorhinal cortex, and its progression to hippocampus and neocortex correlates to the progression of AD symptoms [
311]. Tau is also involved in insulin signaling in the brain, and by the lack of tau protein, hippocampal function and the appetite suppression by insulin in the hypothalamus is weakened which results in metabolic impairments [
60]. Tau does not interact directly with the insulin receptor and IRS-1 but inactivates PI3K/AkT signaling by inhibiting the activity of PTEN (phosphatase and tensin homologue on chromosome 10), which in turn causes hyperphosphorylation of tau [
60,
312].
The ApoE4 gene allele is the strongest genetic risk factor for late-onset AD. Insulin signaling was impaired in an age-dependent manner in ApoE-targeted replacement mice and a fatty diet accelerated this impairment [
313]. By binding with insulin receptor, apoE4 trapped the insulin receptor inside the endosome and interfered with insulin signaling [
313]. This results in decreased mitochondrial respiration and glycolysis, suggesting the role of apoE4 in the pathogenesis in AD in association with insulin resistance.
In summary, decreased glucose metabolism and insulin resistance increase Aβ production by BACE1 activation, deteriorate normal brain functions by the lack of insulin signaling and GSK-3β activation, reduce inhibition of Aβ toxicity and accelerate tau hyperphosphorylation. Additionally, ApoE4 is involved in the insulin signaling process and causes disturbances in energy metabolism.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24043506