 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Martin Jastroch | -- | 2758 | 2023-10-09 16:28:31 | | | |
| 2 | Peter Tang | Meta information modification | 2758 | 2023-10-10 08:07:53 | | |
Video Upload Options
Mitochondria are involved in the regulation of cellular energy metabolism, calcium homeostasis, and apoptosis. For mitochondrial quality control, dynamic processes, such as mitochondrial fission and fusion, are necessary to maintain shape and function. Disturbances of mitochondrial dynamics lead to dysfunctional mitochondria, which contribute to the development and progression of numerous diseases, including Type 2 Diabetes (T2D). Compelling evidence has been put forward that mitochondrial dynamics play a significant role in the metabolism-secretion coupling of pancreatic β cells. The disruption of mitochondrial dynamics is linked to defects in energy production and increased apoptosis, ultimately impairing insulin secretion and β cell death.
1. Introduction: Diabetes—A Global Epidemic
1.1. Glucose Homeostasis
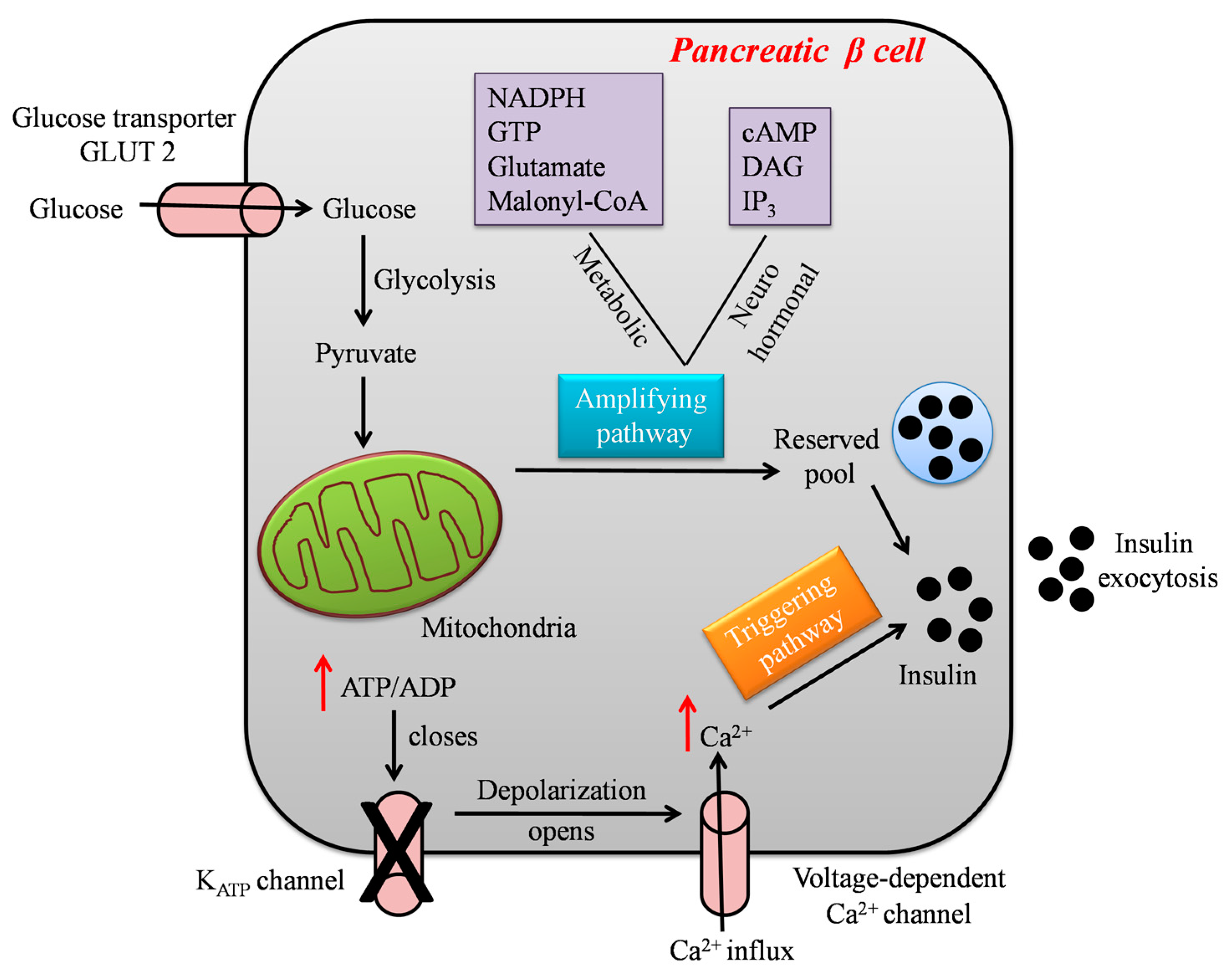
1.2. Glucose-Stimulated Insulin Secretion
2. Mitochondrial Dynamics in Diabetes
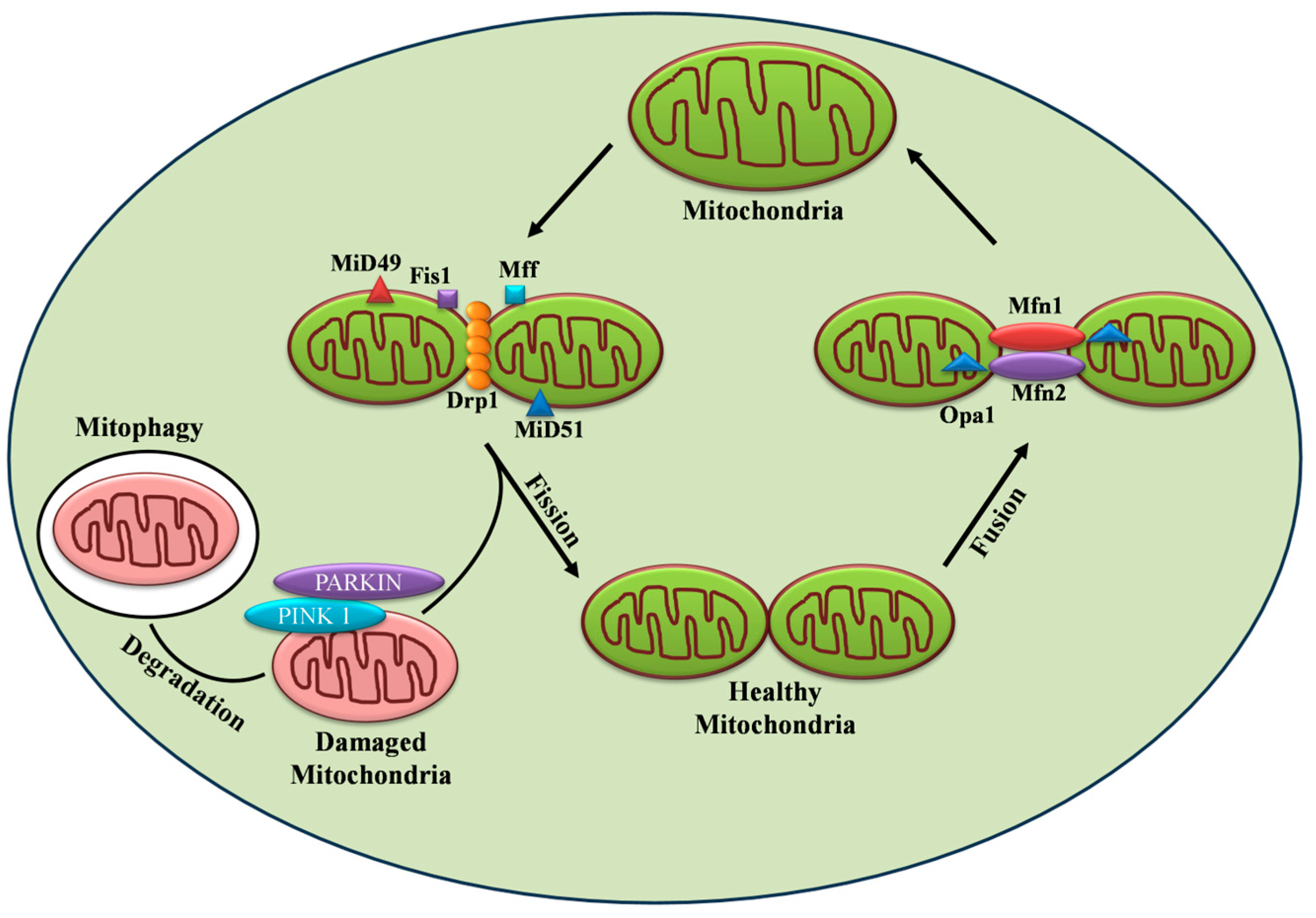
2.1. Mitochondrial Fusion and Its Machinery
2.2. Mitochondrial Fission and Its Machinery
3. Targeting Mitochondrial Dynamic Proteins
References
- Debono, M.; Cachia, E. The impact of diabetes on psychological well being and quality of life. The role of patient education. Psychol. Health Med. 2007, 12, 545–555.
- Chou, C.Y.; Hsu, D.Y.; Chou, C.H. Predicting the Onset of Diabetes with Machine Learning Methods. J. Pers. Med. 2023, 13, 406.
- Baynes, H.W. Classification, pathophysiology, diagnosis and management of diabetes mellitus. J. Diabetes Metab. 2015, 6, 1–9.
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. 2004, 88, 787–835.
- Weyer, C.; Tataranni, P.A.; Bogardus, C.; Pratley, R.E. Insulin resistance and insulin secretory dysfunction are independent predictors of worsening of glucose tolerance during each stage of type 2 diabetes development. Clin. Diabetol. 2001, 2, 167–172.
- Kaiser, N.; Leibowitz, G. Failure of beta-cell adaptation in type 2 diabetes: Lessons from animal models. Front. Biosci.-Landmark. 2009, 14, 1099–1115.
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110.
- Migliorini, A.; Bade, E.; Lickert, H. Islet cell plasticity and regeneration. Mol. Metab. 2014, 3, 268–274.
- Donnelly, R.; Emslie-Smith, A.M.; Gardner, I.D.; Morris, A.D. Vascular complications of diabetes. BMJ 2000, 320, 1062–1066.
- Harding, J.L.; Pavkov, M.E.; Magliano, D.J.; Shaw, J.E.; Gregg, E.W. Global trends in diabetes complications: A review of current evidence. Diabetologia 2019, 62, 3–16.
- De Boer, P.; Giepmans, B.N. State-of-the-art microscopy to understand islets of Langerhans: What to expect next? Immunol. Cell Biol. 2021, 99, 509–520.
- Bosco, D.; Armanet, M.; Morel, P.; Niclauss, N.; Sgroi, A.; Muller, Y.D.; Giovannoni, L.; Parnaud, G.; Berney, T. Unique arrangement of α-and β-cells in human islets of Langerhans. Diabetes 2010, 59, 1202–1210.
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339.
- Cho, J.H.; Kim, J.W.; Shin, J.A.; Shin, J.; Yoon, K.H. β-cell mass in people with type 2 diabetes. J. Diabetes Investig. 2011, 2, 6–17.
- Tups, A.; Benzler, J.; Sergi, D.; Ladyman, S.R.; Williams, L.M. Central regulation of glucose homeostasis. Compr. Physiol. 2011, 7, 741–764.
- Marty, N.; Dallaporta, M.; Thorens, B. Brain glucose sensing, counterregulation, and energy homeostasis. Physiology 2007, 22, 241–251.
- Schwartz, M.W.; Seeley, R.J.; Tschöp, M.H.; Woods, S.C.; Morton, G.J.; Myers, M.G.; D’Alessio, D. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 2013, 503, 59–66.
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53.
- Seino, S. Cell signalling in insulin secretion: The molecular targets of ATP, cAMP and sulfonylurea. Diabetologia 2012, 55, 2096–2108.
- Herman, M.A.; Kahn, B.B. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J. Clin. Investig. 2006, 116, 1767–1775.
- Mertz, R.J.; Worley, J.F.; Spencer, B.; Johnson, J.H.; Dukes, I.D. Activation of Stimulus-Secretion Coupling in Pancreatic β-Cells by Specific Products of Glucose Metabolism: Evidence for privileged signaling by glycolysis. J. Biol. Chem. 1996, 27, 4838–4845.
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261.
- Liemburg-Apers, D.C.; Imamura, H.; Forkink, M.; Nooteboom, M.; Swarts, H.G.; Brock, R.; Smeitink, J.A.; Willems, P.H.; Koopman, W.J. Quantitative glucose and ATP sensing in mammalian cells. Pharm. Res. 2011, 28, 2745–2757.
- Ashcroft, F.M.; Proks, P.; Smith, P.A.; Ämmälä, C.; Bokvist, K.; Rorsman, P. Stimulus–secretion coupling in pancreatic β cells. J. Cell. Biochem. 1994, 55, 54–65.
- Rutter, G.A.; Pullen, T.J.; Hodson, D.J.; Martinez-Sanchez, A. Pancreatic β-cell identity, glucose sensing and the control of insulin secretion. Biochem. J. 2015, 466, 203–218.
- Henquin, J.C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000, 49, 1751–1760.
- Straub, S.G.; Sharp, G.W. Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes/Metab. Res. Rev. 2002, 18, 451–463.
- Kaufman, B.A.; Li, C.; Soleimanpour, S.A. Mitochondrial regulation of β-cell function: Maintaining the momentum for insulin release. Mol. Asp. Med. 2015, 42, 91–104.
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252.
- Wiederkehr, A.; Wollheim, C.B. Impact of mitochondrial calcium on the coupling of metabolism to insulin secretion in the pancreatic β-cell. Cell Calcium 2008, 44, 64–76.
- Kennedy, E.D.; Maechler, P.; Wollheim, C.B. Effects of depletion of mitochondrial DNA in metabolism secretion coupling in INS-1 cells. Diabetes 1998, 47, 374–380.
- De Andrade, P.B.; Rubi, B.; Frigerio, F.; Van den Ouweland, J.M.; Maassen, J.A.; Maechler, P. Diabetes-associated mitochondrial DNA mutation A3243G impairs cellular metabolic pathways necessary for beta cell function. Diabetologia 2006, 49, 1816–1826.
- Silva, J.P.; Köhler, M.; Graff, C.; Oldfors, A.; Magnuson, M.A.; Berggren, P.O.; Larsson, N.G. Impaired insulin secretion and β-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat. Genet. 2000, 26, 336–340.
- Kytövuori, L.; Lipponen, J.; Rusanen, H.; Komulainen, T.; Martikainen, M.H.; Majamaa, K. A novel mutation m. 8561C> G in MT-ATP6/8 causing a mitochondrial syndrome with ataxia, peripheral neuropathy, diabetes mellitus, and hypergonadotropic hypogonadism. J. Neurol. 2016, 263, 2188–2195.
- Dlasková, A.; Špaček, T.; Šantorová, J.; Plecitá-Hlavatá, L.; Berková, Z.; Saudek, F.; Lessard, M.; Bewersdorf, J.; Ježek, P. 4Pi microscopy reveals an impaired three-dimensional mitochondrial network of pancreatic islet β-cells, an experimental model of type-2 diabetes. Biochim. Biophys. Acta-Bioenerg. 2010, 1797, 1327–1341.
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287.
- Newsholme, P.; Haber, E.P.; Hirabara, S.M.; Rebelato, E.L.; Procopio, J.; Morgan, D.; Oliveira-Emilio, H.C.; Carpinelli, A.R.; Curi, R. Diabetes associated cell stress and dysfunction: Role of mitochondrial and non-mitochondrial ROS production and activity. J. Physiol. 2007, 583, 9–24.
- Erion, K.A.; Berdan, C.A.; Burritt, N.E.; Corkey, B.E.; Deeney, J.T. Chronic exposure to excess nutrients left-shifts the concentration dependence of glucose-stimulated insulin secretion in pancreatic β-cells. J. Biol. Chem. 2015, 290, 16191–16201.
- Chang-Chen, K.J.; Mullur, R.; Bernal-Mizrachi, E. β-cell failure as a complication of diabetes. Rev. Endocr. Metab. Disord. 2008, 9, 329–343.
- Peng, L.; Men, X.; Zhang, W.; Wang, H.; Xu, S.; Fang, Q.; Liu, H.; Yang, W.; Lou, J. Involvement of dynamin-related protein 1 in free fatty acid-induced INS-1-derived cell apoptosis. PLoS ONE 2012, 7, e49258.
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2011, 32, 309–319.
- Higa, M.; Zhou, Y.T.; Ravazzola, M.; Baetens, D.; Orci, L.; Unger, R.H. Troglitazone prevents mitochondrial alterations, β cell destruction, and diabetes in obese prediabetic rats. Proc. Natl. Acad. Sci. USA 1999, 96, 11513–11518.
- Mizukami, H.; Wada, R.; Koyama, M.; Takeo, T.; Suga, S.; Wakui, M.; Yagihashi, S. Augmented β cell loss and mitochondrial abnormalities in sucrose-fed GK rats. Virchows Arch. 2008, 452, 383–392.
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005, 48, 282–289.
- Deng, S.; Vatamaniuk, M.; Huang, X.; Doliba, N.; Lian, M.M.; Frank, A.; Velidedeoglu, E.; Desai, N.M.; Koeberlein, B.; Wolf, B.; et al. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes 2004, 53, 624–632.
- Men, X.; Wang, H.; Li, M.; Cai, H.; Xu, S.; Zhang, W.; Xu, Y.; Ye, L.; Yang, W.; Wollheim, C.B.; et al. Dynamin-related protein 1 mediates high glucose induced pancreatic beta cell apoptosis. Int. J. Biochem. Cell Biol. 2009, 41, 879–890.
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99.
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780.
- Benard, G.; Karbowski, M. Mitochondria fusion and fission. Cell Death 2010, 22, 97.
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta (BBA)-Bioenerget. 2008, 1777, 1092–1097.
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065.
- Mohammadi-Motlagh, H.R.; Sadeghalvad, M.; Yavari, N.; Primavera, R.; Soltani, S.; Chetty, S.; Ganguly, A.; Regmi, S.; Fløyel, T.; Kaur, S.; et al. β Cell and Autophagy: What Do We Know? Biomolecules 2023, 13, 649.
- Shan, Z.; Fa, W.H.; Tian, C.R.; Yuan, C.S.; Jie, N. Mitophagy and mitochondrial dynamics in type 2 diabetes mellitus treatment. Aging 2022, 14, 2902.
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645.
- Lee, Y.H.; Kim, J.; Park, K.; Lee, M.S. β-cell autophagy: Mechanism and role in β-cell dysfunction. Mol. Metab. 2019, 27, S92–S103.
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446.
- Jung, H.S.; Chung, K.W.; Kim, J.W.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic β cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324.
- Sarparanta, J.; García-Macia, M.; Singh, R. Autophagy and mitochondria in obesity and type 2 diabetes. Curr. Diabetes Rev. 2017, 13, 352–369.
- Stiles, L.; Shirihai, O.S. Mitochondrial dynamics and morphology in beta-cells. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 725–738.
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015, 524, 309–314.
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121.
- Soleimanpour, S.A.; Gupta, A.; Bakay, M.; Ferrari, A.M.; Groff, D.N.; Fadista, J.; Spruce, L.A.; Kushner, J.A.; Groop, L.; Seeholzer, S.H.; et al. The diabetes susceptibility gene Clec16a regulates mitophagy. Cell 2014, 157, 1577–1590.
- Shi, Y. Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J. Biomed. Res. 2010, 24, 6–15.
- Nicholas, L.M.; Valtat, B.; Medina, A.; Andersson, L.; Abels, M.; Mollet, I.G.; Jain, D.; Eliasson, L.; Wierup, N.; Fex, M.; et al. Mitochondrial transcription factor B2 is essential for mitochondrial and cellular function in pancreatic β-cells. Mol. Metab. 2017, 6, 651–663.
- Pearson, G.L.; Gingerich, M.A.; Walker, E.M.; Biden, T.J.; Soleimanpour, S.A. A selective look at autophagy in pancreatic β-cells. Diabetes 2021, 70, 1229–1241.
- Apostolova, N.; Vezza, T.; Muntane, J.; Rocha, M.; Víctor, V.M. Mitochondrial dysfunction and mitophagy in type 2 diabetes: Pathophysiology and therapeutic targets. Antioxid. Redox Signal. 2023, 39, 278–320.
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.H. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes 2017, 66, 193–205.
- Ding, M.; Dong, Q.; Liu, Z.; Liu, Z.; Qu, Y.; Li, X.; Huo, C.; Jia, X.; Fu, F.; Wang, X. Inhibition of dynamin-related protein 1 protects against myocardial ischemia–reperfusion injury in diabetic mice. Cardiovasc. Diabetol. 2017, 16, 1–11.
- Kabra, U.D.; Pfuhlmann, K.; Migliorini, A.; Keipert, S.; Lamp, D.; Korsgren, O.; Gegg, M.; Woods, S.C.; Pfluger, P.T.; Lickert, H.; et al. Direct Substrate Delivery into Mitochondrial Fission–Deficient Pancreatic Islets Rescues Insulin Secretion. Diabetes 2017, 66, 1247–1257.
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Shealinna, X.G.; et al. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev. Cell. 2017, 40, 583–594.
- Salabei, J.K.; Hill, B.G. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol. 2013, 1, 542–551.
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell. 2006, 10, 839–850.
- Gao, D.; Zhang, L.; Dhillon, R.; Hong, T.T.; Shaw, R.M.; Zhu, J. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS ONE 2013, 8, e60967.
- Qi, X.; Qvit, N.; Su, Y.C.; Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802.
- Guo, X.; Sesaki, H.; Qi, X. Drp1 stabilizes p53 on the mitochondria to trigger necrosis under oxidative stress conditions in vitro and in vivo. Biochem. J. 2014, 461, 137–146.
- Yue, W.; Chen, Z.; Liu, H.; Yan, C.; Chen, M.; Feng, D.; Yan, C.; Wu, H.; Du, L.; Wang, Y.; et al. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 2014, 24, 482–496.


