1. Introduction: Diabetes—A Global Epidemic
Global diabetes mellitus incidences are increasing tremendously in all age groups, affecting the well-being and quality of life of many individuals [
1]. Reports from the International Diabetes Federation stated that nearly 537 million people are suffering from diabetes, which is anticipated to increase to 643 million by 2030 and 783 million by 2045 [
2]. Diabetes is a multifactorial disease, usually associated with persistently high glucose levels (hyperglycemia), either due to impaired insulin secretion or action [
3]. It is categorized into two distinct types: Type 1 (T1D) and Type 2 Diabetes (T2D), wherein T2D accounts for at least 90–95% of total diabetic cases. In people with T1D, also known as early-childhood or juvenile diabetes, the immune system obliterates insulin-producing pancreatic beta (β) cells. In contrast, T2D usually develops later, and the patients are characterized by a blend of two metabolic dysfunctions: insulin resistance and inadequate insulin secretion [
3,
4]. Under conditions of insulin resistance, where insulin-dependent glucose uptake in peripheral organs is reduced, the β cells produce more insulin to compensate, leading to hyperinsulinemia [
5]. Moreover, in some individuals, genetically compromised cells fail to secrete an adequate amount of insulin, resulting in hyperglycemia, the hallmark of T2D. Ultimately, cell mass and functions decrease, further aggravating the pathology [
6]. Whether the exhaustion of β cells is caused by elevated insulin production [
7] or dedifferentiation due to other metabolic complications [
8] remains to be determined. Epidemiological studies reveal that age, lifestyle, ethnicity, smoking, and obesity also contribute to T2D [
4]. Diabetes co-morbidities such as cardiovascular diseases, peripheral vascular diseases, neuropathy, retinopathy, stroke, and nephropathy are the main reasons for mortality in T2D individuals [
9,
10]. Despite extensive research, the pathophysiology related to the progression and complications of T2D is not fully understood. Definitely, to comprehend the underlying mechanisms associated with the disease, it is required to understand the concept of glucose homeostasis (glycemia).
1.1. Glucose Homeostasis
All mammalian cells require glucose as a metabolic substrate for energy production. Thus, it is necessary to sustain adequate levels of blood sugar in the range of 4–7 mmol/L. To maintain normal blood glucose levels, various hormones are released from the brain, liver, adipose tissues, pancreas, intestine, and muscles [
3]. Among these, the endocrine pancreas plays a very critical role in regulating fuel storage by secreting several hormones. Mature endocrine cells, constituting only 2–3% of the total pancreatic volume, aggregate to form a discrete group of cells, which are termed the islets of Langerhans. These pancreatic islets consist of five types of endocrine cells: alpha-cells (15–20%) producing glucagon, beta-cells (65–80%) producing insulin and C-peptide, delta-cells (3–10%) producing somatostatin, gamma cells (3–5%) producing pancreatic polypeptide (PP), and epsilon-cells (<1%) producing ghrelin [
11]. Human islets display a unique architecture wherein all the endocrine cells are bordered by the blood vessels, β cells are mostly located in the center, and α cells occupy the mantle position of the islet. As a result, the ratio of β cells to α cells is relatively higher in the core as compared to the mantle part of the islets [
12]. The highly specialized blood supply pattern through the islet of Langerhans allows the ready exchange of molecules. The mass of β cells is maintained by the differentiation and replication of existing β cells, which are governed by different cell cycle machinery [
13]. The islet cell organization of the diabetic patient and the non-diabetic patient does not differ significantly. The quantitative analysis, however, demonstrated a substantial reduction in β cell mass relative to α cell mass in type 2 diabetic subjects [
14]. Two antagonistic hormones, insulin and glucagon, are crucial in sustaining blood sugar levels in the body [
15]. In response to low blood glucose levels (hypoglycemia), glucagon promotes hepatic glucose release by glycogen breakdown and gluconeogenesis and enhances lipolysis in adipose tissue. In contrast, insulin has a counter-regulatory effect by reducing high blood glucose levels (hyperglycemia). Insulin stimulates glycogen production (glycogenesis) and glucose uptake in the skeletal muscle, liver, and adipose tissues, respectively. Alongside, hepatic glucogenesis and glycogenolysis are decreased, and lipolysis is potently inhibited [
15,
16]. To sense the blood glucose level for regulation of glucose homeostasis, glucose is transported to and metabolized in cells proportionally to the extracellular level [
15,
16,
17].
1.2. Glucose-Stimulated Insulin Secretion
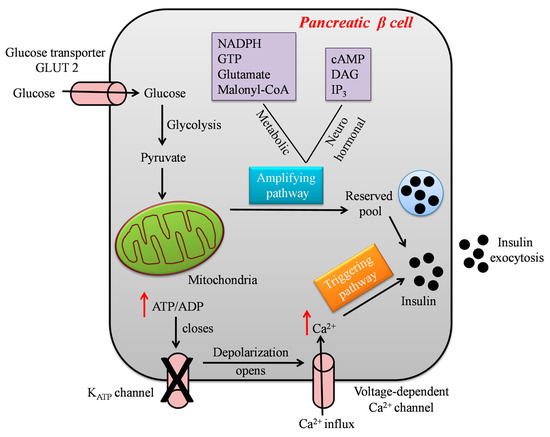
Glucose-Stimulated Insulin Secretion (GSIS) is a complex event modulated by the integration and interactions of multiple signal transductions in β cells [
18]. Although glucose is the chief stimulator of insulin secretion, several hormones, nutrients, neural inputs, chemical messengers, and drugs modulate insulin release [
18,
19]. Following a meal, elevated blood glucose levels lead to glucose uptake via facilitated glucose transport into pancreatic β cells. In the cytoplasm, glucose is metabolized through glycolysis, thereby generating adenosine triphosphate (ATP), pyruvate, nicotinamide adenine dinucleotide (NADH), and water molecules [
20,
21]. The formed pyruvate enters the mitochondrial matrix, and is converted into acetyl-Coenzyme A (CoA), which enters the tricarboxylic acid (TCA) cycle for the generation of the reducing equivalents nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), guanosine triphosphate (GTP), and carbon dioxide (CO
2). The reduced electron carriers NADH and FADH transfer their high-energy electrons to the electron transport chain (ETC), which harvests the energy by step-wise transport to drive protons into the intermembrane space, forming a proton gradient. The proton gradient is used to make ATP by driving the ATP synthase, a process known as oxidative phosphorylation (OXPHOS) [
21,
22,
23]. Specifically, in β cells, the generated ATP elevates the cytoplasmic ATP/ADP ratio, which in turn closes the ATP-sensitive potassium channel, resulting in plasma membrane depolarization. This initiates the opening of L-type voltage-gated calcium channels, facilitating calcium (Ca
2+) ions’ influx into the cells and thereby triggering the exocytosis of insulin granules [
24,
25]. From the above-described sequence of events, it is evident that the mitochondria of pancreatic β cells control GSIS, in particular by transferring the stored energy of glucose to ATP, the main trigger of insulin secretion, as represented in
Figure 1.
Figure 1. A schematic model showing the steps involved in glucose-stimulated insulin secretion by the pancreatic β cell.
GSIS is biphasic and has been considered a combined effect of both triggering and amplifying pathways, as shown in
Figure 1. However, the amplifying pathways are more diverse and detailed as compared to the well-defined triggering pathways [
26]. The triggering pathway initiates the first phase of insulin secretion lasting for 5–10 mins by the K
ATP-dependent mechanism, whereas the second phase of insulin secretion lasts for hours, and the majority of insulin is released in this phase. A sustained second phase is enabled by the amplification pathway, which augments GSIS by K
ATP-independent metabolic signals generated by byproducts of glucose metabolism such as nicotinamide, adenine dinucleotide phosphate hydrogen (NADPH), guanosine triphosphate (GTP), glutamate, and malonyl-CoA [
27]. Several hormones and neurotransmitters acting on membrane receptors regulate the amplification of insulin secretion, such as cyclic adenosine monophosphate (cAMP), diacylglycerol (DAG), and plasma membrane phosphoinositides. In addition to metabolic signals, lipid metabolism also controls and modifies insulin secretion. Glucose-stimulated activation of cell division cycle 42 (Cdc42) pathways is important for second-phase insulin secretion, as it controls the mobilization and exocytosis of insulin. Reports suggest the impairment of amplifying pathways in β cells in both type 2 diabetic animal models and patients. Drugs that can rectify amplifying pathways, e.g., Glucagon-like Peptide 1 (GLP-1) conjugates, could be useful in overriding β cell dysfunction and enhancing insulin secretion in T2D [
26,
27,
28].
2. Mitochondrial Dynamics in Diabetes
Mitochondria are organelles in eukaryotic cells, comprising an inner and an outer membrane separated by the intermembrane space. Beyond their primary role in fueling energy metabolism, they are also involved in several processes including cell signaling, calcium homeostasis, and apoptosis [
29]. The crucial role of mitochondria in metabolic disorders, such as diabetes, is evident from the fact that in β cells, around 80% of glucose oxidation takes place in mitochondria. Therefore, hampering the mitochondrial energy metabolism by blocking the ETC, for example, impairs GSIS [
30]. The significance of mitochondria in metabolism-secretion coupling has further been illustrated in Rho cells devoid of mitochondrial DNA (mtDNA) that no longer respond to glucose. These cells exhibit higher NAD(P)H levels, which in turn inhibit the enzyme glyceraldehyde phosphate dehydrogenase but enhance lactate production by the lactate dehydrogenase enzyme [
31]. The above findings highlight the significance of hydrogen shuttles and mitochondrial respiration in re-oxidizing NAD formed during glycolysis in β cells. The effect of mitochondrial dysfunction is also demonstrated by mtDNA mutations, e.g., the A3243G mutation in a transfer RNA (tRNA), which causes a significant decline in both first- and second-phase insulin secretion [
32]. Further, the knockout of mitochondrial transcription factors A (Tfam) in β cells impair OXPHOS and GSIS, resulting in diabetic mice [
33]. The mutation m.8561C>G in the subunit of mitochondrial ATP synthase (
MT-
ATP6/
8) resulted in impaired complex assembly and decreased ATP production, causing peripheral neuropathy and diabetes mellitus [
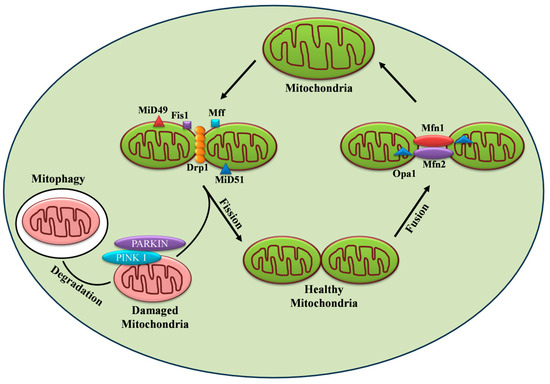
34]. The above findings clearly demonstrate the critical role of mitochondria and their function in insulin secretion. Importantly, mitochondria exist as a dynamic reticular network that frequently undergoes repetitive cycles of fission and fusion in a regulated manner, referred to as “mitochondrial dynamics.” The mitodynamics are regulated by a group of highly conserved dynamin-related GTPases [
29] as shown in
Figure 2. In humans, mice, and rats, β cell mitochondria exist as densely interconnected tubules throughout the cytoplasm [
35]. In the β cell, mitochondrial dynamics compensate for damaged and dysfunctional mitochondria by fusing them with functional ones. On the other hand, fission can drive the removal of damaged or non-functional mitochondria through mitophagy [
36]. It is conceivable to state that a counterbalance between the two dynamic processes is required for normal mitochondrial functionality. Accumulated pieces of evidence support the notion that disturbances in the tuning of the fusion/fission process in the pancreatic β cell led to the development and progression of diabetes in animal and human models.
Figure 2. Regulation of mitochondrial dynamics by fission, fusion, and mitophagy.
Chronic nutrient exposure (e.g., glucose or free fatty acids) has been associated with increased ROS production, mitochondrial dysfunction, and enhanced cell apoptosis, thereby perpetuating diabetes and its complications [
37,
38,
39]. A previous study demonstrated that palmitate-induced mitochondrial fragmentation increased ROS production, lowered ATP production, and increased cell apoptosis [
40]. Similarly, in muscle cells, palmitate treatment caused lipid accumulation, increased oxidative stress, induced mitochondrial fission, and increased insulin resistance [
41]. In prediabetic Zucker diabetic fatty rats, Troglitazone (TZD) treatment protected β cells from lipotoxicity and lipo-apoptosis by enhancing the activity of plasma lipoprotein lipase (LPL), thereby lowering the triglyceride content. TZDs also prevented mitochondrial alteration, improved insulin sensitivity, and henceforth impaired GSIS [
42]. Similarly, β cell mitochondria obtained from diabetic GotoKakizaki rats were found to be disconnected, swollen, and shorter [
43], indicating possible disruption of mitochondrial structure. Islets obtained from type 2 diabetic patients showed disrupted mitochondrial structure, reduced ATP levels, and decreased amounts of insulin granules, with a consequent reduction in insulin secretion [
44,
45]. In INS 1e cells and in islets, hyperglycemia-induced mitochondrial fragmentation decreased membrane potential, increased ROS production, and eventually promoted β cell apoptosis [
46]. Given the evidence for the structure-function relationship of mitochondria, mitochondrial dynamics-regulating proteins are certainly involved in the pathophysiology of diabetes.
2.1. Mitochondrial Fusion and Its Machinery
Mitochondrial fusion is considered the merging of the outer and inner mitochondrial membranes of two different mitochondria to form a larger unit. Fusion enables the rapid exchange of metabolites between neighboring mitochondria, thereby complementing impaired mitochondria and promoting their functionality [
36]. Mitochondrial fusion is also required for the maintenance and distribution of mtDNA. Before entering the S phase of the cell cycle, hyperfusion of mitochondria takes place, thereby increasing ATP production [
36,
47]. The three large dynamin-family GTPases responsible for mitochondrial fusion in mammals are Mitofusins 1 and 2 (Mfn1 and 2) and optic atrophy 1 (Opa1), respectively [
48] as depicted in
Figure 2.
2.2. Mitochondrial Fission and Its Machinery
Mitochondrial fission is defined as the division of mitochondria into two new organelles. Fission is necessary for growing cells to provide them with a sufficient number of mitochondria [
47,
48]. During cellular stress conditions, fission enables the removal of damaged mitochondria and promotes apoptosis, thus helping quality control [
61,
72,
73]. Outer mitochondrial fission is mediated by conserved dynamin family GTPases, mitochondrial fission 1 protein (Fis1), and dynamin-related protein 1 (Drp1) [
48] as shown in
Figure 2.
3. Targeting Mitochondrial Dynamic Proteins
Drp1 also plays a critical role in the regulation of mitophagy as shown in
Figure 2, thus helping to maintain mitochondrial integrity and function necessary for cell survival [
118,
119,
120,
121]. Previous studies also demonstrated mitochondrial fission followed by selective fusion and elimination of dysfunctional mitochondria through mitophagy [
71]. Accumulation of dysfunctional mitochondria and increased ROS production were reported in Type 2 diabetic patients [
122,
123,
124]. In T2D, β cells are exposed to high glucose which is associated with increased oxidative stress and disrupted mitochondrial morphology that hinders the mitophagy pathway and related genes and eventually leads to increased insulin resistance and beta cell death [
119,
122]. Twig et al demonstrated that inhibition of mitochondrial fission by DRP1
K38Aor FIS1 RNAi resulted in decreased mitophagy, reduced mitochondrial respiration, and impaired insulin secretion [
71,
72]. The indispensable molecular players of the mitophagy process are PTEN-induced putative kinase 1 (PINK1), E3 ubiquitin ligase PARKIN and CLEC16A, cardiolipin, transcriptional regulators like Transcription factor B2 (TFB2M), and orphan nuclear receptor Nor1/NR4A3 [
125,
126,
127,
128,
129]. Altered expression of these proteins alters mitochondrial dynamics and impairs the mitophagy process leading to the development of T2D [
130]. The impairment of mitophagy can be restored by mitophagy inducers and can be used as a promising strategy for ameliorating T2D [
119,
131].
The proper functioning of β cell mitochondria is essential due to their involvement in insulin production and release. Unregulated mitochondrial dynamic protein function is associated with numerous pathologies, including T2D. Treatment strategies focusing on manipulating mitochondrial dynamics could possibly ameliorate β cell dysfunction and help maintain glucose homeostasis. Targeting regulators of mitochondrial dynamics in T2D would be a novel approach, but it is very challenging due to multifaceted molecular mechanisms that can lead to unexpected side effects. In recent years, different computational tools have aided in rational drug design and screening of therapeutically important small molecules. These advances have provided breakthroughs for the development of pharmacological compounds that can modulate mitochondrial dynamics. It is widely reported that the compound mdivi-1 inhibits the GTPase activity of Drp1, which is involved in mitochondrial fission. In C2C12 muscle cells, mdivi-1 treatment attenuated palmitic acid-induced mitochondrial fragmentation, oxidative stress, mitochondrial depolarization, and insulin resistance [
41]. Delivery of mdivi-1 into diabetic mice reduced mitochondrial fission, ROS production, inflammation, atherosclerosis, and ameliorated endothelial function [
132]. Mdivi-1 treatment exhibited a cardioprotective effect in HFD-STZ mice by reducing mitochondrial fission, improving mitochondrial function, and suppressing cardiomyocyte apoptosis [
133]. However, a study in the pancreatic β cell line MIN6 and mouse islets demonstrated that mdivi-1 treatment impaired insulin secretion by affecting substrate supply upstream of mitochondria [
113]. Mdivi-1 reversibly inhibits mitochondrial complex I-O
2 consumption and ROS production independently of Drp1 [
114]. Collectively, the short-term potential benefits of mdivi-1 are well demonstrated and can be used in the management of macrovascular complications in T2D. On the other hand, long-term treatment with mdivi-1 suppresses mitochondrial function, decreases mitochondrial mass, and promotes apoptosis in vascular smooth muscles [
134]. Another compound that is a competitive inhibitor of the GTPase activity of Drp1 is Dynasore. It acts by inhibiting the endocytic pathway by blocking coated vesicle formation [
135]. In ischemia/reperfusion mice, dynasore treatment prevented mitochondrial fragmentation and oxidative stress, thereby improving overall cardiac function [
136]. P110, a small peptide inhibitor, blocks the Drp1/Fis1 interaction, which is necessary to dock Drp1 on mitochondria. In cultured neurons, it blocks the binding of Drp1 to Fis1 and exhibits a neuroprotective effect by inhibiting mitochondrial fragmentation and ROS production [
137]. Furthermore, P110 prevented the association of Drp1 with p53 in the ischemia/reperfusion rat and decreased brain infarction and necrotic cell death [
138]. Lastly, 15-Oxospiramilactone (S3), a diterpenoid derivative, inhibits USP30 (mitochondria localized-deubiquitinase) that promotes non-degradative ubiquitination of Mfn1/2, which further enhances Mfn1/2 activity and mitochondrial fusion. In
mfn1–/–or
mfn2–/– knockout cells, it has been observed that S3 restored mitochondrial function and fusion [
139]. In summary, mitochondrial fission inhibitors are promising therapeutic agents for diseases where increased Drp1 expression is involved in the pathology. It is important to note that limited data are available on their effects on metabolic diseases like T2D, and therefore further research is needed. Collectively, however, the application of fission-inhibitors, such as mdivi-1, Dynasore, and P110, or fusion-promoters, such as S3, is not obvious for the treatment of β cell dysfunction, where fission is required during at least the initial phase of GSIS.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241813782