Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dingding Han | -- | 2485 | 2023-10-06 02:36:08 | | | |
| 2 | Rita Xu | Meta information modification | 2485 | 2023-10-07 03:24:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pan, X.; Liu, W.; Du, Q.; Zhang, H.; Han, D. Bacterial Persistence Mechanisms. Encyclopedia. Available online: https://encyclopedia.pub/entry/49855 (accessed on 15 June 2026).
Pan X, Liu W, Du Q, Zhang H, Han D. Bacterial Persistence Mechanisms. Encyclopedia. Available at: https://encyclopedia.pub/entry/49855. Accessed June 15, 2026.
Pan, Xiaozhou, Wenxin Liu, Qingqing Du, Hong Zhang, Dingding Han. "Bacterial Persistence Mechanisms" Encyclopedia, https://encyclopedia.pub/entry/49855 (accessed June 15, 2026).
Pan, X., Liu, W., Du, Q., Zhang, H., & Han, D. (2023, October 06). Bacterial Persistence Mechanisms. In Encyclopedia. https://encyclopedia.pub/entry/49855
Pan, Xiaozhou, et al. "Bacterial Persistence Mechanisms." Encyclopedia. Web. 06 October, 2023.
Copy Citation
The recurrence of bacterial infectious diseases is closely associated with bacterial persisters. This subpopulation of bacteria can escape antibiotic treatment by entering a metabolic status of low activity through various mechanisms, for example, biofilm, toxin–antitoxin modules, the stringent response, and the SOS response.
bacterial persistence
persister formation
single-cell techniques
1. Introduction
In 1944, Joseph Bigger reported a small number of survivors, namely persisters, for the first time when he was studying staphylococcus treated with intermittent penicillin sterilization. Unlike resistant strains, descendants of persisters are susceptible to penicillin, suggesting that survival ability is inheritable [1]. Since then, an increasing number of reports have indicated that persisters are associated with relapsing infections and can survive therapy in diseases such as tuberculosis and urinary tract infections, despite the susceptibility to antibiotics seen in clinical laboratory tests [2]. Persister is a bacteria subpopulation that grows slowly or in a dormant manner. The state of persistence can be induced by a variety of mechanisms, and their antibiotic susceptibility is generally reduced due to dormancy [3]. This feature of persisters makes them unable to be completely eliminated during antibiotic treatment, which increases the likelihood of resistance and can lead to infection recurrence [4]. It is generally believed that persisters are crucial for pathogens to survive antimicrobial chemotherapy when the host’s immune response is limited [5]. The incidence of persister infections is especially high in immunocompromised patients with the human immunodeficiency virus or those undergoing cancer chemotherapy. For example, increased numbers of persister cells of Pseudomonas aeruginosa (P. aeruginosa) are a major concern for immunocompromised and cystic fibrosis patients, leading to high morbidity and mortality [6]. However, in immunocompetent individuals in cases where the pathogen is located at sites poorly accessible by components of the immune system, persister formation is also likely to take place [5]. Consequently, the site of infection would play a role in persister formation.
With the studies of persistence, a few intracellular bacteria have recently been found to exhibit a persistent phenotype [7]. One of the most distinctive features of persistence is the biphasic killing curve resulting from the coexistence of antibiotic-susceptible and persistent cells [8]. After being exposed to antibiotics, the most susceptible bacteria are rapidly killed, while a small proportion of persisters survive longer in a non-growing state (Figure 1). Once antibiotics are removed, persisters recover to grow and exhibit similar patterns to antibiotic-susceptible bacteria [9][10]. Besides antibiotics, some other stress environments, such as acid, toxic metals, and high temperatures, can also induce persister formation [11]. Nonetheless, antibiotic-induced persistence is of more concern in the hospital setting. In the clinic, antibiotic treatment is usually administered at regular dosages and times. Therefore, the antibiotic concentration in vivo fluctuates periodically [12]. In long-term treatment patients who received periodic antibiotics of a high concentration, the proportion of persisters in isolated P. aeruginosa was 100 times higher than during the early stages of treatment. However, the minimum inhibitory concentration (MIC) did not change significantly [13]. Accordingly, the failure of antibiotic treatment may not be solely due to resistance, as persistence may also play a role.
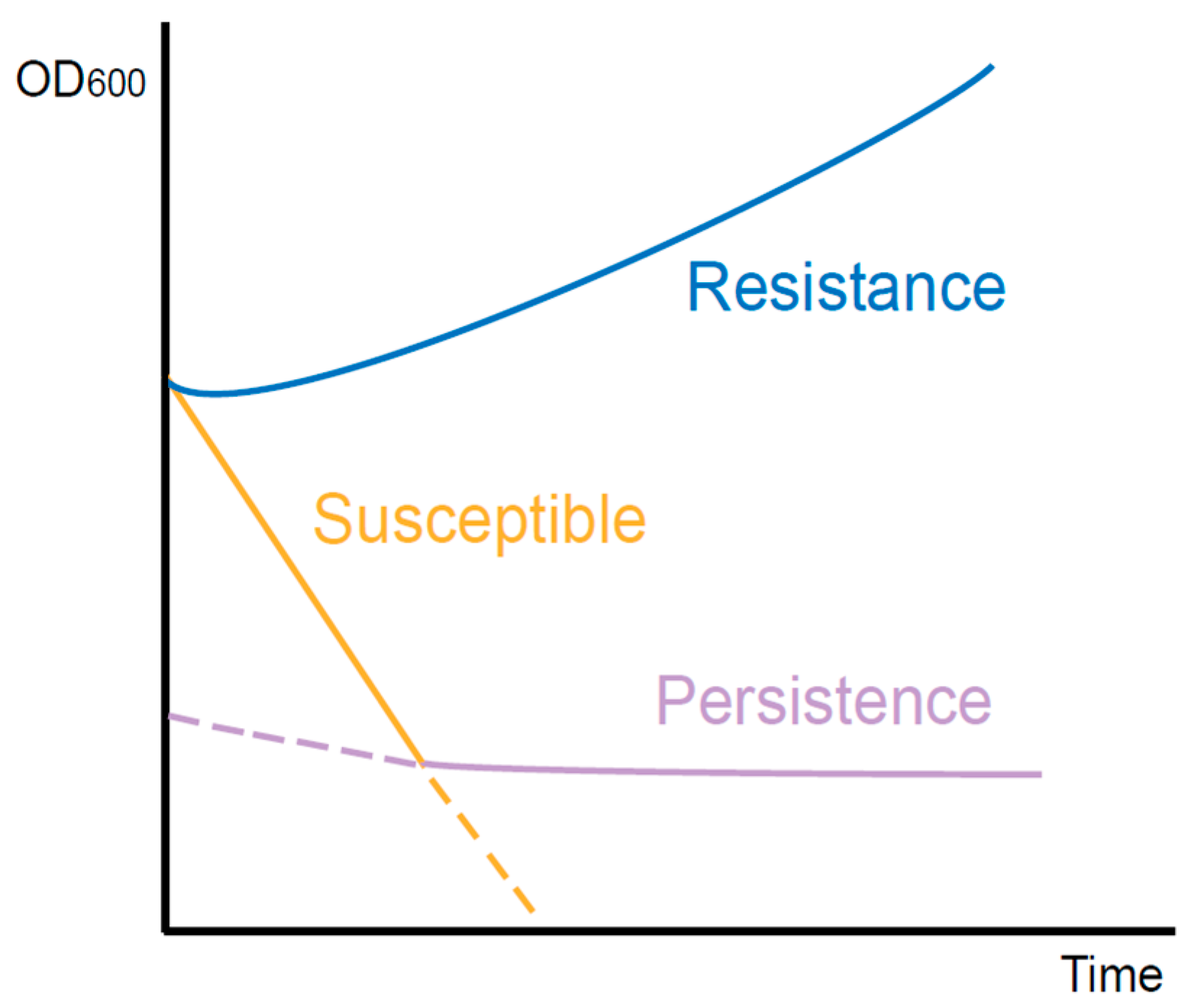
Figure 1. Killing curves of different types of bacteria during antibiotic exposure. The resistant bacteria continue to grow. The susceptible bacteria are killed quickly. However, with the existence of persisters, the curve declines slowly.
2. The Difference between Persistence and Resistance
Resistance to antibiotics is generally inherited through genetic mutations in bacteria. Despite high antibiotic concentrations, these resistant cells are still capable of dividing. The mechanisms of resistance have been deeply studied by researchers worldwide and can be categorized into three common pathways [14]. Firstly, bacteria reduce the intracellular antibiotic concentration in the way of either activating efflux pumps or decreasing the permeability of their membranes. Secondly, they actively modify the intracellular targets of antibiotics, thereby avoiding antibiotic attacks. Furthermore, by generating specific enzymes, antibiotics are directly inactivated within bacterial cells. As opposed to resistance, persistence is a heterogeneous population behavior. The vast majority of the population presents susceptibility to the stress, which is not impacted by the non-growth persister component. This susceptible feature can remain stable over generations, suggesting that all bacteria within the population have the same genetic background as persisters. Therefore, antibiotic persistence is a phenotypic change that occurs in a small proportion of cells. Compared to the stable and heritable resistance of antibiotic resisters, the ability of persisters to survive lethal antibiotics is transient and reversible [15].
Although the bacterial population containing persisters exhibited antimicrobial susceptibility, the killing curve of surviving persisters was independent of antibiotic concentration. However, the mortality rate of antibiotic-resistant bacteria was associated with antibiotic concentration when this was higher than the MIC [4]. Additionally, resistance is often specific to a certain class of drug, while persistence responds to a wide range of stimuli, including antibiotics. This reflects the differences in the underlying molecular mechanisms of the two species.
3. Research on the Mechanism of Bacterial Persistence
3.1. Biofilm
Persistence and biofilm formation have been shown to be closely related in numerous studies [16][17][18]. Bacteria attach irreversibly to the surface of inert or active entities, secrete proteins and polysaccharide matrix, and form a kind of membrane by wrapping the bacterial colonies. This membrane has strong selective permeability to nutritional and antibacterial compounds [19]. As a result, the biofilm provides shelter for potential persisters to escape antibiotics and immune molecules [12]. Existing studies have mainly emphasized the physical protection effect of biofilm. However, increasing evidence indicates that hypoxia and low nutrition in the inner biofilm also contribute to persister formation [20]. The tricarboxylic acid cycle would be downregulated in persisters inside the biofilm when exposed to antibiotic-induced oxidative stress, thereby preventing the production of reactive oxygen species, an essential component of antibiotic-induced bacterial killing [21]. A real-time impedance-based technology was used to examine the kinetics of P. aeruginosa biofilm growth under regular antibiotics [22]. The results showed that biofilm formation rate and outcome varied between different culture media. Although antibiotics can delay biofilm growth and reduce its amount, biofilm formation cannot be completely inhibited at a low antibiotic concentration. Based on these findings, a ‘fortress model’ of central persisters’ growth in the biofilm was proposed [23] (Figure 2). Nonetheless, bacteria at the periphery of the biofilm can form stronger biofilms that are resistant to antibiotics due to their easier access to nutrients than those growing in the core of the biofilm [24][25]. These studies suggest that nutrition gradients may influence the persister. However, it is still controversial whether growing at the periphery or core of the biofilm facilitates persister formation. Furthermore, the deficiency of the capsule can lead to the biofilm formation. It has been reported that in carbapenem-resistant Klebsiella pneumoniae (K. pneumoniae), the mutants that have deletions of core capsule biosynthesis genes produce more robust biofilm compared with the wild type, enhancing epithelial cell invasion and persistence in urinary tract infections [26].
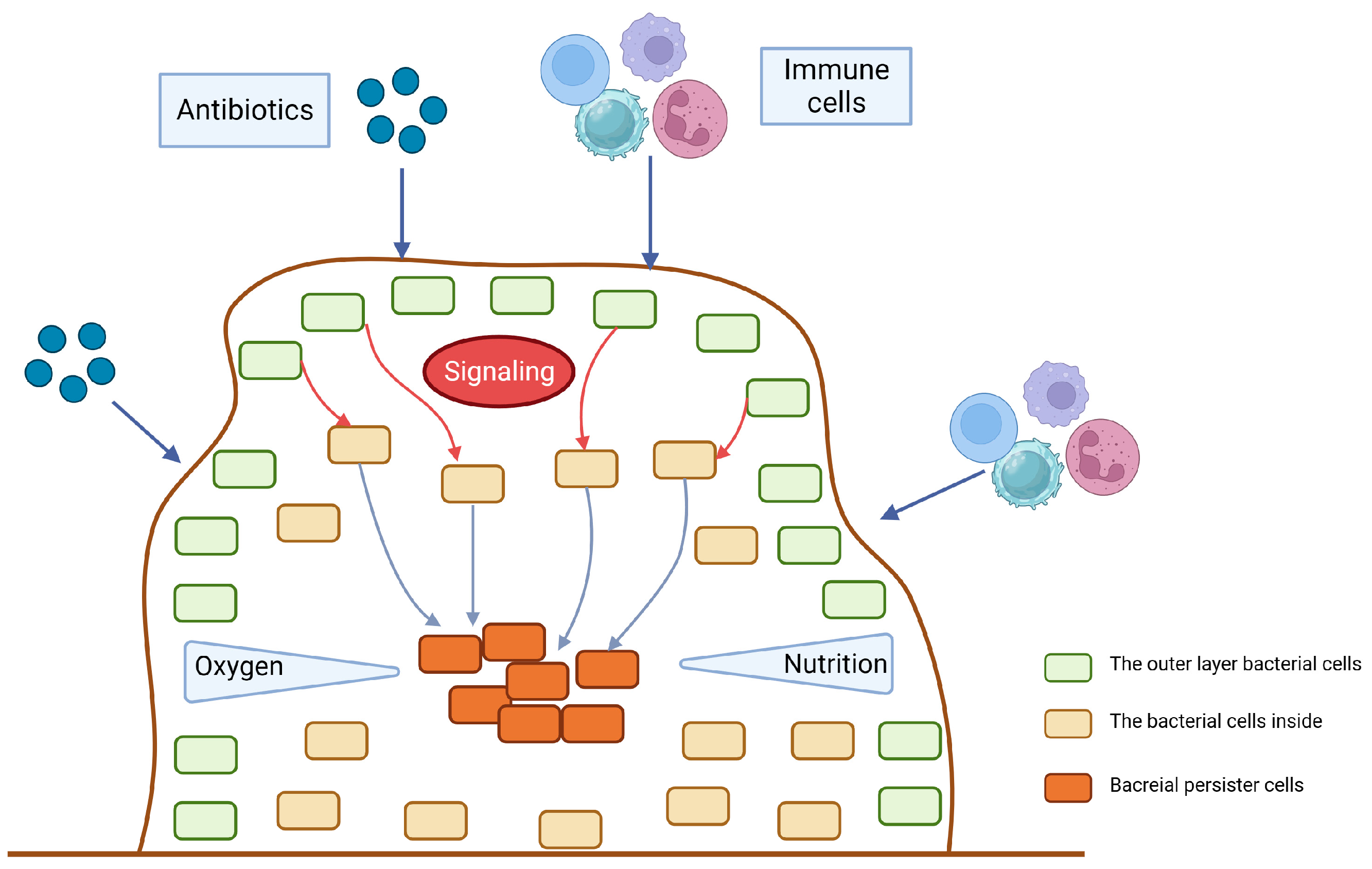
Figure 2. The fortress model. When antibiotics, immune cells or other potential environment stress start to stimulate the surface of the biofilm, the outer layer of bacteria will send signals to the bacteria inside to be alert to the bactericidal attack. The gradient of nutrition and oxygen will also contribute to the persister dormancy.
3.2. Toxin–Antitoxin Modules
Bacterial toxin–antitoxin (TA) modules extensively exist in a variety of cellular processes [27], and are composed of a toxin protein that inhibits cell growth by interfering with important metabolic activities and an antitoxin that can protect cells from toxins [3][28]. Some experiments have demonstrated that breaking the balance between the toxin and antitoxin would lead bacterial growth to a stagnant state, and then weaken the harm of antibiotics or other unfavorable circumstances, which could facilitate the survival of bacteria [13][29]. Environmental stress can induce a graded and controlled activation of toxins to help bacteria enter a transient dormancy state, which is the basis for surviving high-concentration antibiotics [30]. For example, the stress inside the host macrophages is more severe than the nutritious culture medium, providing more triggers for the TA modules’ activation [2]. To date, TA systems can be divided into eight types based on the properties and working modes of antitoxin, named type I to type VIII, following the discovery order. Type I antitoxins are small RNAs that inhibit the cognate toxin’s expression by silencing their transcript. Type II antitoxins are proteins that neutralize the cognate toxin by binding to them and forming tight complexes [31]. While type I TA modules were the first to be discovered, type II are the most abundant and diverse, occurring in the majority of bacteria and some archaea. Since the discovery of type I and type II TA modules on bacterial chromosomes, they have been recognized as beneficial factors in the face of stressful conditions including antibiotic persistence [17]. For type I TA systems, high persistence was reported to be positively correlated with the levels of ObgE and HokB. Obg is a conserved GTPase that plays an important role at the crossroads of the major cellular processes of translation and DNA replication, which are considered to be repressed in dormant persister cells. HokB, a toxin encoded by a type I toxin–antitoxin module, can provoke a collapse in the membrane potential [32]. The gene high persistence protein A (hipA)-encoding type II toxin was firstly discovered in Escherichia coli (E. coli), whose corresponding antitoxin is HipB [33][34]. It was reported that strains carrying hipA7 mutants produce persisters at a frequency of 1% compared to 10-5 in wild-type strains when exposed to ampicillin [35]. As the bacterial cell density increases, hipA7 strains also produce persistent cells at an increasing frequency. In addition, the deficiencies of more than five type II TA modules in E. coli would decrease persister formation during the exponential phase [36]. Beside the mutations on TA module components, the expression of TA modules in enriched E. coli persisters was obviously upregulated compared with normal growing bacteria [33][36]. Furthermore, the Lon protease, a kind of ATPase in E. coli, is required for the degradation of the antitoxin, and results in the accumulation of toxins to promote persister formation [29].
Current studies have been focused on Gram-negative bacteria, especially E. coli, though the study on Gram-positive bacteria is not clear. There is evidence that the absence of the TA module in Staphylococcus aureus (S. aureus) would not affect the persister level [37]. Nonetheless, it was proposed that TA modules in Gram-positive bacteria work similarly to those in Gram-negative bacteria, and take part in the host’s stress response [38].
3.3. ppGpp and Stringent Response
If cells encountered amino acid starvation, protein synthesis and other metabolic activities would be closed. This phenomenon is called the stringent response, a type of conservative and global transcriptomic adaptation to environmental pressure [39]. Many studies have confirmed that the stringent response can facilitate persister formation by lapsing into dormancy [7][40][41]. The small molecule guanosine tetraphosphate (ppGpp), controlled by Rel/Spo homolog and small alarmone synthetase proteins, is the central molecule in the stringent response [41][42]. In many biological and animal models, the bacterial mutants that cannot produce ppGpp usually show a reduced ability of persister formation [25][43][44][45]. In the stationary phase, the elevated recruitment of ppGpp to Obg GTPase results in the excessive binding of Obg to the 50S ribosome subunit, which reduces the levels of active 70S ribosomes and thereby regulates cellular functions and participates in various stress adaptions like the stringent response [32][46]. In persisters, Obg induces the expression of HokB, a membrane-targeted type I toxin, which can damage proton motion and block ATP synthesis, leading to dormancy [32]. Recent evidence has put an emphasis on ppGpp instead of TA modules in persister formation [47]. The steady state of ppGpp is maintained by RelA and SpoT, while the former is a synthase, and the latter is a hydrolase with weak synthetic activity [48][49]. It has been proven that ciprofloxacin, cefepime, colistin, and amikacin can upregulate the expression of the relA gene to help persister formation [50]. However, without the presence of ppGpp, such as relA knockout mutants, persisters still exist [51], suggesting that the mechanism needs further investigation. PhoU is another persistence-related factor. PhoU homologs have been identified as phosphate-specific transport system accessory proteins that respond to the environmental Pi level. Deletion of the PhoU1 and PhoU2 in S. aureus caused an increase in both persistence and bacterial virulence [52]. In P. aeruginosa, a phoU mutant showed increased levels of intracellular ppGpp, affected antibiotic susceptibility, and decreased growth rate [53]. Interestingly, biofilm formation was not affected by the phoU mutation.
During the stringent response, the accumulation of ppGpp results in changes in multiple metabolic pathways, involving DNA replication, nucleotide synthesis, transcription, ribosome maturation, and lipid metabolism. For example, the stringent response decreases the initiation rate of DNA replication in E. coli [54], and controls 100S ribosome formation by transcriptional regulation in multiple species [55]. rRNA synthesis and genes involved in the metabolism of macromolecules, such as phospholipids, are generally repressed, while the transcription of nutrient transporters is increased to overcome nutrient limitations [56]. In addition, due to the decreased level of lysine during fatty acid starvation, RelA is distinctly activated and induces the synthesis of ppGpp [57].
To explore the mechanism of how the stringent response increases bacterial cytotoxicity, Thomas A Hooven et al. conducted transposon sequencing and RNA sequencing analyses on Streptococcus agalactiae (GBS) with human whole blood [39]. Except in the capsular polysaccharide genes, relA was identified to be essential for GBS survival in blood. In addition to promoting the persistence of GBS in host blood, the activation of a stringent response by relA can also enhance the expression of β-hemolysin/cytolysin, a toxin required for GBS virulence [58] through an arginine-mediated metabolism pathway. At present, a small molecule inhibitor aimed at the stringent response has been developed as a new type of antibacterial, and it has demonstrated the corresponding therapeutic effects in pre-clinical tests [59][60].
3.4. SOS Response
The SOS response plays an important role in DNA repair, which is controlled by two key factors: repressor factor LexA and inducible factor RecA [61]. RecA protein is a recombinase which is important for repairing bacterial recombinational DNA [62]. The failure of damage repair by the SOS response usually leads to cell death, while successful DNA repair may eliminate the lysis function of antibiotics [63][64]. High cell density within biofilms, together with oxidative stress, triggers DNA damage and evokes the SOS response [61]. For instance, Salmonella persisters could retain the function of initiating infection recurrence via RecA-mediated DNA repair [65]. In response to the stress changes, RecA mediates global transcription by stimulating the self-lysis of LexA [62][66]. In addition, the SOS response is proven to enhance the expression of fibronectin, helping biofilm formation [67]. For resuscitation experiments, it was also observed that after the removal of ofloxacin, the SOS response continuously increased in persistent E. coli cells [68] (Figure 3).
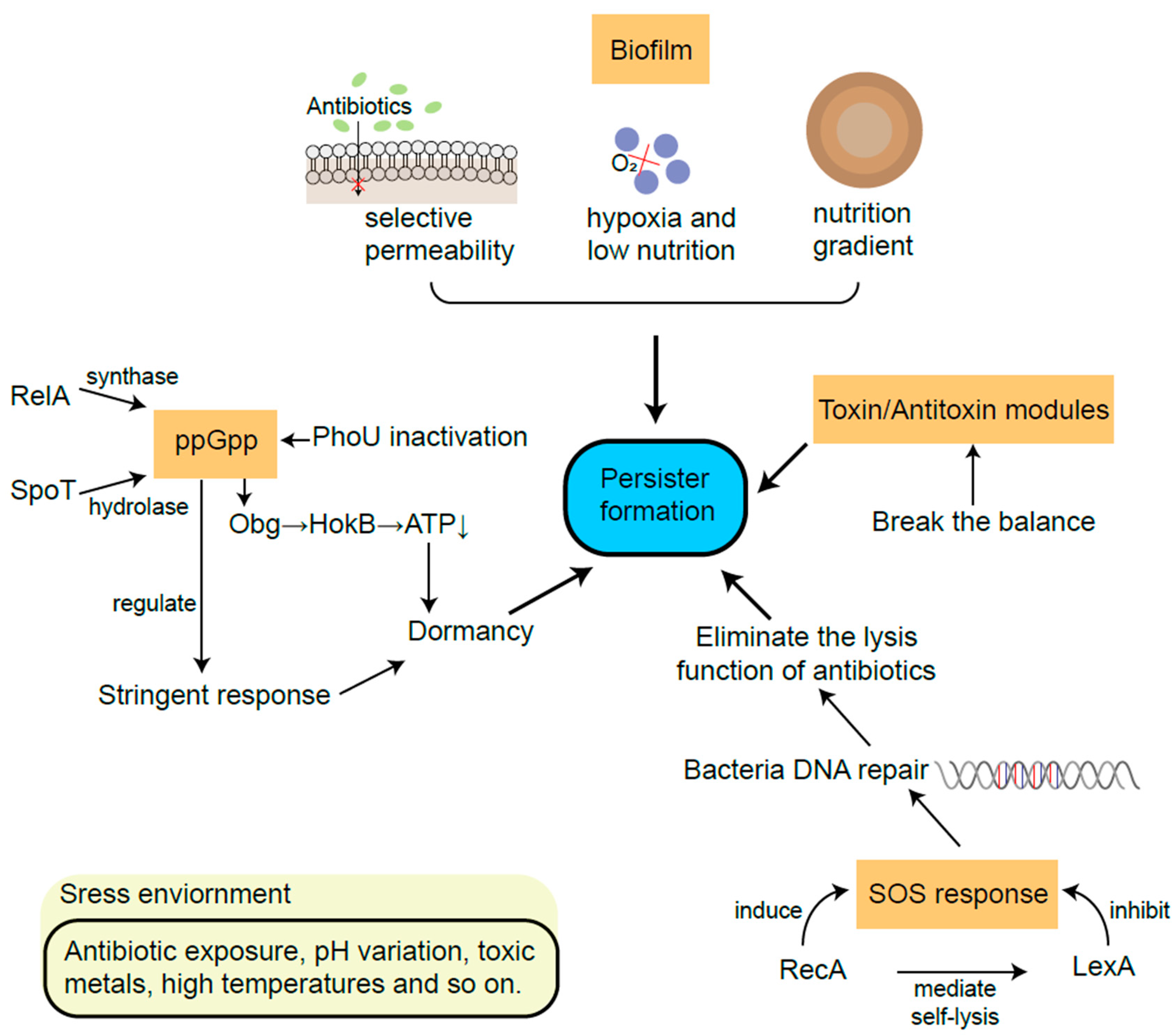
Figure 3. The main mechanisms of persister formation from biofilm, TA modules, ppGpp and SOS response.
Of the mechanisms described above, biofilm has been demonstrated in vivo in human pathology. For example, the biofilm formation of Mycobacterium tuberculosis in human lungs has been indicated to contribute to drug tolerance in experiments in animal models of infection and in the lung tissues of patients [69]. However, TA modules, the stringent response, and the SOS response have not been well elucidated in the physiopathology of specific diseases despite their documentation in vitro at the moment.
References
- Bigger, J.W. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 1944, 244, 497–500.
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464.
- Liu, Y.; Yang, J.; Zhao, Z.; Pu, Y.; Bai, F. Bacterial persistence. Sci. China Chem. 2014, 57, 1625–1633.
- Gollan, B.; Grabe, G.; Michaux, C.; Helaine, S. Bacterial Persisters and Infection: Past, Present, and Progressing. Annu. Rev. Microbiol. 2019, 73, 359–385.
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372.
- Moradali, M.F.; Ghods, S.; Rehm, B.H. Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell Infect. Microbiol. 2017, 7, 39.
- Peyrusson, F.; Varet, H.; Nguyen, T.K.; Legendre, R.; Sismeiro, O.; Coppee, J.Y.; Wolz, C.; Tenson, T.; Van Bambeke, F. Intracellular Staphylococcus aureus persisters upon antibiotic exposure. Nat. Commun. 2020, 11, 2200.
- Balaban, N.Q.; Helaine, S.; Lewis, K.; Ackermann, M.; Aldridge, B.; Andersson, D.I.; Brynildsen, M.P.; Bumann, D.; Camilli, A.; Collins, J.J.; et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 2019, 17, 441–448.
- Kim, J.S.; Yamasaki, R.; Song, S.; Zhang, W.; Wood, T.K. Single cell observations show persister cells wake based on ribosome content. Environ. Microbiol. 2018, 20, 2085–2098.
- Rollin, G.; Tan, X.; Tros, F.; Dupuis, M.; Nassif, X.; Charbit, A.; Coureuil, M. Intracellular Survival of Staphylococcus aureus in Endothelial Cells: A Matter of Growth or Persistence. Front. Microbiol. 2017, 8, 1354.
- Harms, A.; Maisonneuve, E.; Gerdes, K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science 2016, 354, aaf4268.
- Yan, J.; Bassler, B.L. Surviving as a Community: Antibiotic Tolerance and Persistence in Bacterial Biofilms. Cell Host Microbe 2019, 26, 15–21.
- Cui, P.; Xu, T.; Zhang, W.H.; Zhang, Y. Molecular mechanisms of bacterial persistence and phenotypic antibiotic resistance. Yi Chuan 2016, 38, 859–871.
- Blair, J.M.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51.
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 2016, 14, 320–330.
- Beroz, F.; Yan, J.; Sabass, B.; Stone, H.A.; Bassler, B.L.; Wingreen, N.S.; Meir, Y. Verticalization of bacterial biofilms. Nat. Phys. 2018, 14, 954–960.
- Helaine, S.; Cheverton, A.M.; Watson, K.G.; Faure, L.M.; Matthews, S.A.; Holden, D.W. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 2014, 343, 204–208.
- Burmolle, M.; Thomsen, T.R.; Fazli, M.; Dige, I.; Christensen, L.; Homoe, P.; Tvede, M.; Nyvad, B.; Tolker-Nielsen, T.; Givskov, M.; et al. Biofilms in chronic infections—A matter of opportunity—Monospecies biofilms in multispecies infections. FEMS Immunol. Med. Microbiol. 2010, 59, 324–336.
- Cendra, M.D.M.; Blanco-Cabra, N.; Pedraz, L.; Torrents, E. Optimal environmental and culture conditions allow the in vitro coexistence of Pseudomonas aeruginosa and Staphylococcus aureus in stable biofilms. Sci. Rep. 2019, 9, 16284.
- Conlon, B.P. Staphylococcus aureus chronic and relapsing infections: Evidence of a role for persister cells: An investigation of persister cells, their formation and their role in S. aureus disease. Bioessays 2014, 36, 991–996.
- Van Acker, H.; Van Dijck, P.; Coenye, T. Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends Microbiol. 2014, 22, 326–333.
- Ziemyte, M.; Carda-Dieguez, M.; Rodriguez-Diaz, J.C.; Ventero, M.P.; Mira, A.; Ferrer, M.D. Real-time monitoring of Pseudomonas aeruginosa biofilm growth dynamics and persister cells’ eradication. Emerg. Microbes Infect. 2021, 10, 2062–2075.
- Lewis, K. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 2001, 45, 999–1007.
- Uzoechi, S.C.; Abu-Lail, N.I. Variations in the Morphology, Mechanics and Adhesion of Persister and Resister E. coli Cells in Response to Ampicillin: AFM Study. Antibiotics 2020, 9, 235.
- Nguyen, D.; Joshi-Datar, A.; Lepine, F.; Bauerle, E.; Olakanmi, O.; Beer, K.; McKay, G.; Siehnel, R.; Schafhauser, J.; Wang, Y.; et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 2011, 334, 982–986.
- Ernst, C.M.; Braxton, J.R.; Rodriguez-Osorio, C.A.; Zagieboylo, A.P.; Li, L.; Pironti, A.; Manson, A.L.; Nair, A.V.; Benson, M.; Cummins, K.; et al. Adaptive evolution of virulence and persistence in carbapenem-resistant Klebsiella pneumoniae. Nat. Med. 2020, 26, 705–711.
- Shao, Y.; Harrison, E.M.; Bi, D.; Tai, C.; He, X.; Ou, H.Y.; Rajakumar, K.; Deng, Z. TADB: A web-based resource for Type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res. 2011, 39, D606–D611.
- Hayes, F.; Kedzierska, B. Regulating toxin-antitoxin expression: Controlled detonation of intracellular molecular timebombs. Toxins 2014, 6, 337–358.
- Maisonneuve, E.; Shakespeare, L.J.; Jorgensen, M.G.; Gerdes, K. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. USA 2011, 108, 13206–13211.
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, Targets, and Triggers: An Overview of Toxin-Antitoxin Biology. Mol. Cell 2018, 70, 768–784.
- Jurenas, D.; Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Biology and evolution of bacterial toxin-antitoxin systems. Nat. Rev. Microbiol. 2022, 20, 335–350.
- Verstraeten, N.; Knapen, W.J.; Kint, C.I.; Liebens, V.; Van den Bergh, B.; Dewachter, L.; Michiels, J.E.; Fu, Q.; David, C.C.; Fierro, A.C.; et al. Obg and Membrane Depolarization Are Part of a Microbial Bet-Hedging Strategy that Leads to Antibiotic Tolerance. Mol. Cell 2015, 59, 9–21.
- Keren, I.; Shah, D.; Spoering, A.; Kaldalu, N.; Lewis, K. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 2004, 186, 8172–8180.
- Schumacher, M.A.; Piro, K.M.; Xu, W.; Hansen, S.; Lewis, K.; Brennan, R.G. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 2009, 323, 396–401.
- Korch, S.B.; Henderson, T.A.; Hill, T.M. Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol. Microbiol. 2003, 50, 1199–1213.
- Shah, D.; Zhang, Z.; Khodursky, A.; Kaldalu, N.; Kurg, K.; Lewis, K. Persisters: A distinct physiological state of E. coli. BMC Microbiol. 2006, 6, 53.
- Conlon, B.P.; Rowe, S.E.; Gandt, A.B.; Nuxoll, A.S.; Donegan, N.P.; Zalis, E.A.; Clair, G.; Adkins, J.N.; Cheung, A.L.; Lewis, K. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 2016, 1, 16051.
- Donegan, N.P.; Cheung, A.L. Regulation of the mazEF toxin-antitoxin module in Staphylococcus aureus and its impact on sigB expression. J. Bacteriol. 2009, 191, 2795–2805.
- Hooven, T.A.; Catomeris, A.J.; Bonakdar, M.; Tallon, L.J.; Santana-Cruz, I.; Ott, S.; Daugherty, S.C.; Tettelin, H.; Ratner, A.J. The Streptococcus agalactiae Stringent Response Enhances Virulence and Persistence in Human Blood. Infect. Immun. 2018, 86, e00612-17.
- Patel, H.; Buchad, H.; Gajjar, D. Pseudomonas aeruginosa persister cell formation upon antibiotic exposure in planktonic and biofilm state. Sci. Rep. 2022, 12, 16151.
- Boutte, C.C.; Crosson, S. Bacterial lifestyle shapes stringent response activation. Trends Microbiol. 2013, 21, 174–180.
- Tozawa, Y.; Nomura, Y. Signalling by the global regulatory molecule ppGpp in bacteria and chloroplasts of land plants. Plant Biol. (Stuttg) 2011, 13, 699–709.
- Viducic, D.; Ono, T.; Murakami, K.; Susilowati, H.; Kayama, S.; Hirota, K.; Miyake, Y. Functional analysis of spoT, relA and dksA genes on quinolone tolerance in Pseudomonas aeruginosa under nongrowing condition. Microbiol. Immunol. 2006, 50, 349–357.
- Maisonneuve, E.; Castro-Camargo, M.; Gerdes, K. (p)ppGpp Controls Bacterial Persistence by Stochastic Induction of Toxin-Antitoxin Activity. Cell 2018, 172, 1135.
- Fung, D.K.; Chan, E.W.; Chin, M.L.; Chan, R.C. Delineation of a bacterial starvation stress response network which can mediate antibiotic tolerance development. Antimicrob. Agents Chemother. 2010, 54, 1082–1093.
- Chakraborty, A.; Halder, S.; Kishore, P.; Saha, D.; Saha, S.; Sikder, K.; Basu, A. The structure-function analysis of Obg-like GTPase proteins along the evolutionary tree from bacteria to humans. Genes. Cells 2022, 27, 469–481.
- Svenningsen, M.S.; Veress, A.; Harms, A.; Mitarai, N.; Semsey, S. Birth and Resuscitation of (p)ppGpp Induced Antibiotic Tolerant Persister Cells. Sci. Rep. 2019, 9, 6056.
- Si, L.; Gu, J.; Wen, M.; Wang, R.; Fleming, J.; Li, J.; Xu, J.; Bi, L.; Deng, J. relA Inactivation Converts Sulfonamides Into Bactericidal Compounds. Front. Microbiol. 2021, 12, 698468.
- Atkinson, G.C.; Tenson, T.; Hauryliuk, V. The RelA/SpoT homolog (RSH) superfamily: Distribution and functional evolution of ppGpp synthetases and hydrolases across the tree of life. PLoS ONE 2011, 6, e23479.
- Baek, M.S.; Chung, E.S.; Jung, D.S.; Ko, K.S. Effect of colistin-based antibiotic combinations on the eradication of persister cells in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2020, 75, 917–924.
- Chowdhury, N.; Kwan, B.W.; Wood, T.K. Persistence Increases in the Absence of the Alarmone Guanosine Tetraphosphate by Reducing Cell Growth. Sci. Rep. 2016, 6, 20519.
- Shang, Y.; Wang, X.; Chen, Z.; Lyu, Z.; Lin, Z.; Zheng, J.; Wu, Y.; Deng, Q.; Yu, Z.; Zhang, Y.; et al. Staphylococcus aureus PhoU Homologs Regulate Persister Formation and Virulence. Front. Microbiol. 2020, 11, 865.
- de Almeida, L.G.; Ortiz, J.H.; Schneider, R.P.; Spira, B. phoU inactivation in Pseudomonas aeruginosa enhances accumulation of ppGpp and polyphosphate. Appl. Environ. Microbiol. 2015, 81, 3006–3015.
- Ferullo, D.J.; Lovett, S.T. The stringent response and cell cycle arrest in Escherichia coli. PLoS Genet. 2008, 4, e1000300.
- Prossliner, T.; Skovbo Winther, K.; Sorensen, M.A.; Gerdes, K. Ribosome Hibernation. Annu. Rev. Genet. 2018, 52, 321–348.
- Krasny, L.; Gourse, R.L. An alternative strategy for bacterial ribosome synthesis: Bacillus subtilis rRNA transcription regulation. EMBO J. 2004, 23, 4473–4483.
- Geiger, T.; Goerke, C.; Fritz, M.; Schafer, T.; Ohlsen, K.; Liebeke, M.; Lalk, M.; Wolz, C. Role of the (p)ppGpp synthase RSH, a RelA/SpoT homolog, in stringent response and virulence of Staphylococcus aureus. Infect. Immun. 2010, 78, 1873–1883.
- Shahi, I.; Llaneras, C.N.; Perelman, S.S.; Torres, V.J.; Ratner, A.J. Genome-Wide CRISPR-Cas9 Screen Does Not Identify Host Factors Modulating Streptococcus agalactiae beta-Hemolysin/Cytolysin-Induced Cell Death. Microbiol. Spectr. 2022, 10, e0218621.
- Andresen, L.; Varik, V.; Tozawa, Y.; Jimmy, S.; Lindberg, S.; Tenson, T.; Hauryliuk, V. Auxotrophy-based High Throughput Screening assay for the identification of Bacillus subtilis stringent response inhibitors. Sci. Rep. 2016, 6, 35824.
- Wexselblatt, E.; Oppenheimer-Shaanan, Y.; Kaspy, I.; London, N.; Schueler-Furman, O.; Yavin, E.; Glaser, G.; Katzhendler, J.; Ben-Yehuda, S. Relacin, a novel antibacterial agent targeting the Stringent Response. PLoS Pathog. 2012, 8, e1002925.
- Podlesek, Z.; Zgur Bertok, D. The DNA Damage Inducible SOS Response Is a Key Player in the Generation of Bacterial Persister Cells and Population Wide Tolerance. Front. Microbiol. 2020, 11, 1785.
- Cox, M.M. Regulation of bacterial RecA protein function. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 41–63.
- Theodore, A.; Lewis, K.; Vulic, M. Tolerance of Escherichia coli to fluoroquinolone antibiotics depends on specific components of the SOS response pathway. Genetics 2013, 195, 1265–1276.
- Guerin, E.; Cambray, G.; Sanchez-Alberola, N.; Campoy, S.; Erill, I.; Da Re, S.; Gonzalez-Zorn, B.; Barbe, J.; Ploy, M.C.; Mazel, D. The SOS response controls integron recombination. Science 2009, 324, 1034.
- Hill, P.W.S.; Moldoveanu, A.L.; Sargen, M.; Ronneau, S.; Glegola-Madejska, I.; Beetham, C.; Fisher, R.A.; Helaine, S. The vulnerable versatility of Salmonella antibiotic persisters during infection. Cell Host Microbe 2021, 29, 1757–1773.
- Little, J.W. Mechanism of specific LexA cleavage: Autodigestion and the role of RecA coprotease. Biochimie 1991, 73, 411–421.
- McCourt, J.; O’Halloran, D.P.; McCarthy, H.; O’Gara, J.P.; Geoghegan, J.A. Fibronectin-binding proteins are required for biofilm formation by community-associated methicillin-resistant Staphylococcus aureus strain LAC. FEMS Microbiol. Lett. 2014, 353, 157–164.
- Goormaghtigh, F.; Van Melderen, L. Single-cell imaging and characterization of Escherichia coli persister cells to ofloxacin in exponential cultures. Sci. Adv. 2019, 5, eaav9462.
- Chakraborty, P.; Bajeli, S.; Kaushal, D.; Radotra, B.D.; Kumar, A. Biofilm formation in the lung contributes to virulence and drug tolerance of Mycobacterium tuberculosis. Nat. Commun. 2021, 12, 1606.
More
Information
Subjects:
Microbiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
561
Revisions:
2 times
(View History)
Update Date:
07 Oct 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No