Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Razvan Adrian Covache-Busuioc | -- | 2914 | 2023-09-25 11:58:11 | | | |
| 2 | Peter Tang | Meta information modification | 2914 | 2023-09-26 02:58:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Toader, C.; Eva, L.; Covache-Busuioc, R.; Costin, H.P.; Glavan, L.; Corlatescu, A.D.; Ciurea, A.V. Multifaceted Role of Golgi Apparatus. Encyclopedia. Available online: https://encyclopedia.pub/entry/49595 (accessed on 10 August 2026).
Toader C, Eva L, Covache-Busuioc R, Costin HP, Glavan L, Corlatescu AD, et al. Multifaceted Role of Golgi Apparatus. Encyclopedia. Available at: https://encyclopedia.pub/entry/49595. Accessed August 10, 2026.
Toader, Corneliu, Lucian Eva, Razvan-Adrian Covache-Busuioc, Horia Petre Costin, Luca-Andrei Glavan, Antonio Daniel Corlatescu, Alexandru Vlad Ciurea. "Multifaceted Role of Golgi Apparatus" Encyclopedia, https://encyclopedia.pub/entry/49595 (accessed August 10, 2026).
Toader, C., Eva, L., Covache-Busuioc, R., Costin, H.P., Glavan, L., Corlatescu, A.D., & Ciurea, A.V. (2023, September 25). Multifaceted Role of Golgi Apparatus. In Encyclopedia. https://encyclopedia.pub/entry/49595
Toader, Corneliu, et al. "Multifaceted Role of Golgi Apparatus." Encyclopedia. Web. 25 September, 2023.
Copy Citation
Functioning as the cellular “sorting hub”, the Golgi apparatus is indispensable in protein categorization and adjustments crucial to the dynamic equilibrium of neuronal structure, function, and synaptic adaptability. The Golgi apparatus is not only pivotal in protein categorization and alterations but also stands out as a central orchestrator of neuronal maturation.
Golgi apparatus
neuronal plasticity
neuronal development
neurogenesis
Alzheimer’s disease
SARS-CoV-2
1. Introduction
The complexity of the nervous system’s architecture is underpinned by processes of neuronal maturation and synaptic adaptability. Central to these intricate mechanisms is an oft-overlooked cellular entity in neurobiological discussions: the Golgi apparatus. Functioning as the cellular “sorting hub”, the Golgi apparatus is indispensable in protein categorization and adjustments crucial to the dynamic equilibrium of neuronal structure, function, and synaptic adaptability [1]. Contemporary research has illuminated potential associations between the Golgi apparatus and the pathogenesis of Alzheimer’s, particularly emphasizing the role of the Golgi matrix protein GM130 (a morphological determinant protein situated on the cis-face of the Golgi apparatus) [2][3][4]. Novel insights are surfacing, linking this protein to its Golgi-based counterpart and the pathological shifts characteristic of Alzheimer’s, thus ushering in innovative investigative trajectories in our pursuit of understanding this devastating neurodegenerative disorder [4].
The Golgi apparatus is not only pivotal in protein categorization and alterations but also stands out as a central orchestrator of neuronal maturation. The nuanced orchestration of axonal and dendritic differentiation is facilitated by this organelle, paving the way for growth and distinctiveness within neural circuits. Moreover, its influence is unmistakably evident in synaptic plasticity, fundamental processes governing learning and memory within the nervous system [5].
2. Golgi Apparatus in Axonal Development
Cerebral cells, comprising neurons and glia, are characterized by intricate architectures tailored to execute designated roles. Neurons establish synapses to facilitate electrical communication, oligodendrocytes envelop axons with myelin sheaths for insulating properties, and astrocytes bridge vascular networks [6][7]. These specialized morphologies necessitate distinct routes for protein trafficking, prominently featuring the Golgi apparatus outposts (GOPs) (Figure 1). In specialized cellular contexts, Golgi outposts assume critical functions essential for the sculpting of distinct cellular morphologies and architectures. Notably, within neurons, these organelles are pivotal for the formation of dendritic branches [8]. Concurrently, in muscle cells, they are implicated in the genesis of grid-like microtubule lattices, and in oligodendrocytes, they facilitate the formation of microtubules that encircle the myelin sheath in a spiral configuration [9]. Intriguingly, notwithstanding their crucial roles, to date, Golgi outposts have been exclusively identified in vivo and within primary cultured cells, evading detection in immortalized cell lines [10].
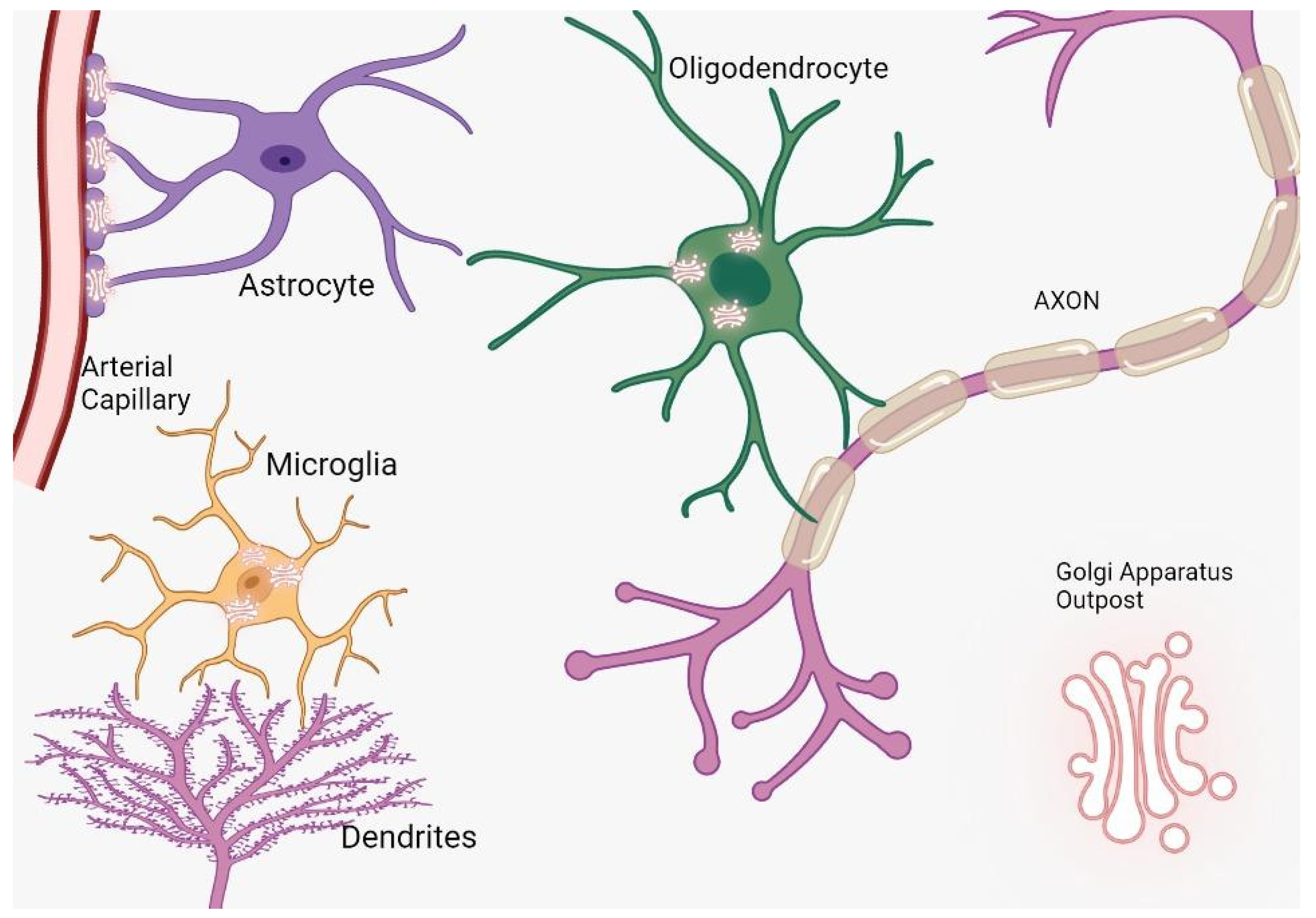
Figure 1. The distribution and function of Golgi outposts in different types of glial cells. In oligodendrocytes, these outposts are located in processes and the myelin sheath, where they nucleate new microtubules along radial and lamellar microtubules. In microglia, Golgi outposts contribute to the nucleation of new microtubules and the establishment of branched processes. In astrocytes, Golgi outposts are observed in endfeet contacting blood vessels as evidenced by electron microscopy, although their specific function is yet to be determined.
In summary, the secretory pathway initiates in the endoplasmic reticulum (ER), where nascent proteins are synthesized. Subsequently, these proteins traverse through the Golgi apparatus (GA) for further maturation, processing, and sorting. They are then dispatched to subsequent post-Golgi/trans-Golgi network (TGN) compartments, ultimately reaching their designated locations such as endosomes, lysosomes, and the plasma membrane [11]. Transmembrane proteins synthesized on the RER are incorporated into the ER membrane subsequently exiting through specialized sites termed ERES (ER exit site) with the aid of COPII vesicles [12]. Following the detachment of COPII proteins [13], these vesicles employ SNAREs (Soluble NSF Attachment Protein Receptors) to amalgamate with tubular assemblies termed ER-Golgi intermediate compartments (ERGICs). Additionally, certain proteins can retrogress from the ER via COPI vesicles initiated at the Golgi apparatus. Within the ERGIC, proteins undergo processing and subsequently navigate to the Golgi apparatus for further modifications, encompassing glycosylation and proteolysis. This journey encompasses the cis-Golgi network (CGN), medial Golgi cisternae, and the trans-Golgi network (TGN) [14].
Upon reaching the TGN, proteins adopt diverse processing trajectories contingent upon their intended function and localization. Vesicles emanating from the Golgi, laden with secreted or transmembrane proteins, can bifurcate and merge with plasma membranes, undergoing subsequent processing. This can culminate in the extracellular secretion of proteins or their integration into plasma membranes. Additionally, vesicles laden with secretory proteins can be conserved proximal to plasma membranes or endosomes, awaiting release upon specific stimuli. Moreover, proteins destined for endosomes and lysosomes undergo modification via an oligosaccharide marker termed mannose 6-phosphate, and subsequently depart the TGN enclosed within clathrin-coated vesicles [15]. Consequently, the selection of secretory routes is determined by the intended role, ultimate destination, and inherent nature of the respective cargo proteins [16].
3. Golgi Apparatus Involvement in Dendritic Formation
Neuronal maturation is characterized by the augmentation of the plasma membrane during processes such as polarization and outgrowth. The localization of membrane constituents is contingent upon the secretory pathway, a sequence encompassing a myriad of organelles such as the endoplasmic reticulum (ER), ER-Golgi intermediate compartment, cis-Golgi, medial Golgi, and trans-Golgi, among others [17]. These specialized organelles orchestrate the synthesis and delivery of novel membrane lipids and proteins; however, the understanding of their spatial arrangement, functionality, and regulatory mechanisms within neurons remains nascent.
In non-neuronal cells, the architecture of the secretory pathway organelles is notably conserved. The ER permeates the cell and includes distinct locales termed “ER exit sites”. Here, the coat protein complex II-coated vesicles, laden with newly synthesized cargo, commence their journey to the Golgi complex for subsequent protein processing and sorting. Ultimately, post-Golgi carriers facilitate the conveyance to the plasma membrane [18][19].
The elaboration and extension of dendrites and axons potentially necessitate coordinated modifications in cytoskeletal organization and membrane transport mechanisms. Specifically, molecules such as RhoA [20] and MAP2 [21] appear to be predisposed to augment dendritic growth. Concurrently, a commensurate adaptation in membrane provisioning might be requisite to facilitate their distinctive morphologies.
Intriguingly, neurons also harbor unique secretory conduits in dendrites, which encompass both ER and Golgi outposts [16]. Yet, a conspicuous absence of Golgi outposts in axons prompts inquiries regarding the potential role of the polarized distribution of Golgi proteins in delineating the differential maturation trajectories of dendrites and axons. Contemporary research intimates a proactive involvement of secretory trafficking in the genesis of specialized dendritic compartments. Nevertheless, the extent to which these secretory pathways modulate the differential elongation patterns of dendrites and axons remains enigmatic (as illustrated in Figure 2). Furthermore, the implications of dendritic Golgi outposts in dendritic elaboration, and their putative role in sculpting the distinct morphological attributes of dendrites and axons, await elucidation [22].
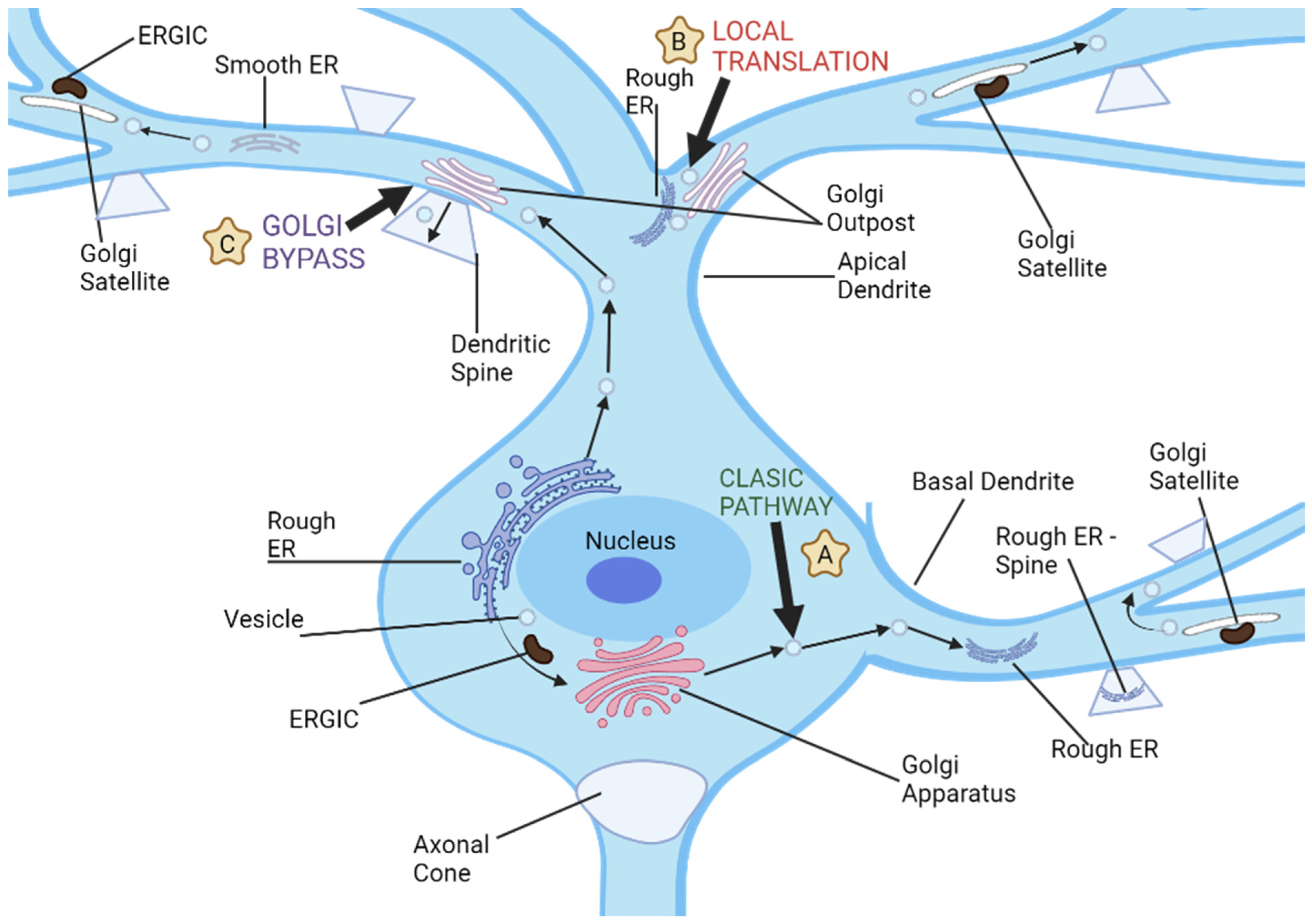
Figure 2. Neuronal dendrites provide multiple pathways for cargo transport and secretory processes, and there are three distinct routes used for dendritic cargo transportation. (A) The classic secretion pathway involves protein translation in neuronal cell bodies, followed by exit from endoplasmic reticulum and further processing by the ER-Golgi intermediate compartments and cell body Golgi. Vesicles then transport these proteins to dendrites or other parts of a neuron for storage and transport. (B) Local dendritic translation involves synthesizing proteins on dendritic rough ER before sending them off for further modification by dendritic Golgi outposts. After leaving Golgi outposts, proteins are transported via post-Golgi vesicles which may either travel along dendrites or fuse with either dendritic plasma membranes or synaptic spines. (C) Golgi bypass pathway allows cargo synthesized on the cell body ER to bypass its Golgi organelle and be transported directly to dendrites for modification by Golgi outposts before being packaged into post-Golgi vesicles destined for synapses or plasma membrane.
Pertaining to GOP genesis, a plausible mechanism might encompass localized de novo synthesis from the ER, reminiscent of the Golgi reconstitution observed at cellular extremities in non-neuronal entities [23]. Alternatively, GOPs could emerge post somatic Golgi fragmentation due to heightened neuronal activity, with remnants of the degraded Golgi potentially serving as blueprints for the subsequent assembly of satellite Golgi arrays [24]. Formation of GOPs might also ensue from the primary somatic GA, prompting inquiries into the participation of Golgi fission machinery components such as LIMK1 [25] and Protein Kinase D1 (PKD1) [26], and the potential involvement of their proximal regulators and distal effectors. Notably, evidence underscores the potential influence of a RhoA-Rho kinase (Rock) signaling cascade in the orchestration of polarized GOPs during neuronal morphogenesis.
The Golgi apparatus inherently harbors the capability to act as a reservoir of membrane constituents vital for localized cellular expansion [22]. Dendrites replete with Golgi elements consistently manifest greater length and intricate architecture as compared to their Golgi-devoid counterparts. Empirical assessments have ascertained that an overwhelming 86% of neurons under examination showcased Golgi elements within their most elongated dendrite [27].
In mature neurons, a hallmark characteristic resides in their intricately branched dendritic arbors, which conspicuously surpass the complexity of dendrites devoid of Golgi outposts.
Collectively, these revelations accentuate that a non-uniform Golgi distribution resonates with dendritic expansion patterns. The enhanced length and complexity of Golgi-endowed dendrites underscore the indispensable role of this organelle in sculpting dendritic architecture.
4. BARS Regulation of Golgi Trafficking
The CtBP (C-terminal-binding protein) family, comprising CtBP1 and CtBP2, is pivotal in a gamut of biological undertakings such as development, differentiation, oncogenesis, and apoptosis, chiefly functioning as transcriptional co-repressors within the nuclear compartment [28]. BARS (Brefeldin A ADP-Ribosylated Substrate) emerges as a salient mediator in membrane fission activities including macropinocytosis, fluid-phase endocytosis, COPI-coated vesicle genesis, and an array of post-Golgi carrier and ribbon events during mitosis. The intricacies of BARS in fission are well-chronicled in cellular studies, where it is posited as a key intermediary in basolateral post-Golgi carrier formation. Specifically, MDCK cells, manifesting polarized epithelial attributes, display hindered Vesicular Stomatitis Virus G (VSVG) transport upon BARS inhibition, attributed to tubular carriers laden with this payload remaining adhered to the Trans Golgi Network (TGN). This fission process orchestrated by BARS at the TGN entails a multifaceted ensemble of proteins [29][30].
Through in situ hybridization evaluations, it is apparent that CtBP family constituents are profusely expressed across the neural framework. Notably, CtBP1 and CtBP2 showcase divergent distribution spectra within mature cerebral structures, distinguished by expression magnitudes, territorial expression blueprints, subcellular targeting proficiencies, and intracellular positioning paradigms. Mouse-based investigations with deletions in one or both CtBP genes intimate the existence of both common and unique functionalities for these proteins [31][32]. Of particular significance is the discernment that concurrent excision of CtBP1 and CtBP2 in murine models precipitates retarded development of forebrain and midbrain structures, a sequela of modulated transcription factor activities in their absence [33][34].
Diminished BARS expression is correlated with marked curtailment in hippocampal neuronal growth, migration, and the multipolar-to-bipolar transitional dynamics in cortical neurons. Notably, these effects can be mitigated by co-expression of fission-inefficient mutants exhibiting reduced nuclear positioning or fission vigor. Contrarily, accentuated growth is observed upon co-expression of the said mutants, an effect which can be counteracted by the concurrent expression of aforementioned mutants [30].
The meticulous orchestration of membrane constituents along the secretory trajectory is cardinal for neuronal tasks such as augmentation, polarity inception, sustenance, synaptic plasticity, and cellular migration [35][36]. This conduit is initiated at the rough endoplasmic reticulum, subsequently traversing the Golgi machinery where membrane proteins designated for axons and dendrites are segregated into specific carriers for extracellular dispatch [37]. Upon departure from the trans-Golgi matrix, they are relayed to plasma membranes via molecular propellants, exemplified by kinesin superfamily entities and myosins. Despite this knowledge, the specialized apparatus governing their TGN exit remains inadequately delineated. Nevertheless, it is established that BARS, an integral affiliate of the CtBP assemblage, is indispensable during neuronal ontogeny [38].
Both LIMK1 and PKD1 are quintessential components of the TGN fission apparatus in polarized epithelial structures, overseeing membrane protein transference either to apical or basolateral facets [39][40]. Concurrently, these proteins, evident within neurons situated at GAs, partake in dynamin-facilitated fission of Golgi conduits, thereby engendering Golgi outposts. Ensuing research ought to delve into the mechanisms by which BARS engenders dendritic carrier fissions at TGNs and probe the putative role of BARS in the fission of Golgi conduits during the generation of Golgi outposts [41].
5. Golgi Apparatus and Synaptic Plasticity
Rare diseases, particularly genetic anomalies, exert a pronounced effect on the nervous system, culminating in manifestations such as neurodegeneration and behavioral perturbations [42]. These infrequent neurological conditions furnish a platform to elucidate hitherto uncharted cellular dynamics integral to neuronal performance. This is especially pertinent in disorders associated with ATP7A/ATP7B genes and COG complex subunit genes. The proteins expressed from these genes predominantly localize within the Golgi apparatus under standard conditions [43]. Over the course of evolution, organisms have intricately modulated copper metabolism and transport. Central to this regulatory framework are ATP7A and ATP7B proteins. These proteins, classified under P-type Cu-transporting ATPases, harness ATP hydrolysis to facilitate the transmembrane movement of copper ions. Essentially, these copper pumps either extrude surplus copper from cells or allocate it to copper-reliant enzymes. The physiological roles of Cu-transporting ATPases are multifaceted, spanning dietary copper excretion through bile, placental transport, and lactational secretion to modulating resistance against select anti-cancer therapeutics [44][45][46][47].
Mutational alterations in the ATP7A gene underpin Menkes disease, typified by systemic copper deficiency stemming from hindered intestinal copper assimilation [48]. The clinical picture of Menkes disease predominantly emerges in childhood, exhibiting a spectrum of systemic and neurologically linked symptoms. The latter include intellectual impairments and gray matter neurodegeneration [49]. Such clinical manifestations may be ascribed to perturbations in copper-reliant enzymes that traverse the Golgi apparatus or reside within mitochondria [50]. Paradoxically, cell-specific aberrations in ATP7A precipitate intracellular copper accumulation, a scenario arising despite the overarching copper deficiency evident in Menkes patients [51]. In a contrasting paradigm, mutations in the ATP7B gene underlie Wilson’s disease. This disorder is marked by hepatic impediments to copper excretion, ensuing in systemic copper excess, hepatotoxicity, psychiatric manifestations, and neurodegeneration, particularly in the lenticular domain [50].
Differential expressions of ATP7 and COG complex subunits can significantly modulate synaptic morphology, mitochondrial constituents, and neurotransmission in response to stimulatory or plasticity-inducing cues. A deeper exploration is imperative to elucidate these interconnected dynamics. Mitochondria, pivotal cellular organelles, mediate synaptic operations through diverse modalities. They serve as calcium reservoirs [52], facilitate ATP synthesis [53], generate Krebs cycle intermediates influencing neurotransmission [54], and engage in glutamate metabolism [55]. Additionally, mitochondria confer metabolic adaptability by diversifying carbon inputs into the Krebs cycle, including sources such as pyruvate, glutamate, or fatty acids [55].
The mitochondria’s competence in modulating synaptic calcium concentrations might shed light on certain neurotransmission phenotypes, particularly in contexts of ATP7 overexpression or disrupted copper efflux. Brief episodes of high-frequency neuronal excitation can engender synaptic plasticity via facilitation, augmentation, and potentiation, cumulatively enhancing transmission efficiency, termed synaptic enhancement [56]. Facilitation hinges on the preservation of optimal calcium concentrations post-influx, while concurrently augmenting it with basal calcium during neuronal activity and inducing exocytosis of readily releasable vesicles in response to neural excitation [57][58][59]. In contrast, potentiation seeks a balance between exocytosis and endocytosis [60].
Post-tetanic facilitation and augmentation typically involve a diminution in residual calcium [56]. Basal calcium concentrations appear cardinal for post-tetanic potentiation (PTP), with activity-mediated calcium elevations instigating phosphorylation-oriented signaling persisting beyond its ephemeral phase [60]. Given the involvement of mitochondria during PTP evolution, their potential role here merits consideration [61][62].
Mitochondria’s indispensable role in modulating calcium concentrations at neuronal terminals is paramount for synaptic plasticity and the inception of short-term memories. They act as calcium buffers postconditioning [63]. However, in synapses manifesting ATP7 overexpression or compromised copper efflux pathways, any enhancements post-sustained tetanic or post-tetanic stimuli are negated. It is salient that upon diminishing COG complex expression, both mitochondrial content and neurotransmission phenotypes in ATP7 hyperexpressing terminals are concurrently ameliorated, substantiating the hypothesis of mitochondria-mediated mechanisms dictating these phenotypes [64][65].
6. LARGE Gene Interactions with Golgi Apparatus—Consequences and Implications
The LARGE gene, prominently expressed in the hippocampus compared to other tissues, plays a pivotal role in cognitive functions [52]. Mutations within this gene give rise to human congenital muscular dystrophy type 1D, characterized by severe cognitive impairments, atypical electroretinogram results, and nuanced structural cerebral anomalies [53][54]. LARGEmyd mice, harboring truncation mutations of the LARGE gene, exhibit neurological manifestations akin to humans bearing such mutations, including sensorineural hearing loss, retinal transmission deficits, neurodevelopmental irregularities, and attenuated long-term potentiation [LTP] [55][56]. Recent research underscores the potential of aberrant synaptic operations as the underlying cause of such cognitive impediments [55].
Intellectual disability, historically termed mental retardation, is delineated by pronounced cognitive (IQ ≤ 70) and adaptive behavior deficits, manifested as restricted conceptual, social, or practical skills prior to 18 years of age [66]. This disability might also correlate with other cognitive deteriorations across one’s lifetime, including dementia of a neurodegenerative origin. Numerous X-linked cerebral disorders related to intellectual disability have proteins, instrumental in synaptic signaling pathways, as plausible etiological agents [67][68][69]. Fragile X syndrome, a predominant genetically inherited intellectual disability, is directly associated with AMPA-R dysregulation. Proteins PAK3 and OPHN1, implicated in intellectual disability, oversee synaptic AMPA-R expression and stability [65]. Mutations in its GluA3 subunit have been identified in individuals with X-linked intellectual disability [70]. However, the specifics of these perturbations in AMPA-R dynamics and their contribution to cognitive dysfunction remain largely enigmatic.
The LARGE gene is instrumental in regulating AMPA-R—a synaptic receptor crucial for synaptic plasticity and long-term potentiation, vital for memory consolidation and learning. Mice devoid of this protein manifest compromised LTP, potentially correlated with neurodevelopmental aberrations such as anomalous neuronal migratory patterns [71].
Microscopic evaluations have identified dystroglycan in the hippocampus, particularly within postsynaptic assemblies crafted by mossy fibers on pyramidal neurons [72]. Prior investigations revealed that dystroglycan suppression, achieved using GFAP-Cre, results in diminished LTP, proposing its role in synaptic functionality [73]. Synaptic adaptability alterations might arise due to diminished dystroglycan in either neurons or glial cells—fundamental to the orchestration of synaptic operations [74]. The potential contribution of developmental cerebral anomalies in influencing the electrophysiological attributes of the GFAP-Cre/DG-null brain cannot be dismissed [55].
References
- Liu, J.; He, J.; Huang, Y.; Xiao, H.; Jiang, Z.; Hu, Z. The Golgi apparatus in neurorestoration. J. Neurorestoratol. 2019, 7, 116–128.
- Liu, C.; Mei, M.; Li, Q.; Roboti, P.; Pang, Q.; Ying, Z.; Gao, F.; Lowe, M.; Bao, S. Loss of the golgin GM130 causes Golgi disruption, Purkinje neuron loss, and ataxia in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 346–351.
- Elsherbini, A.; Zhu, Z.; Quadri, Z.; Crivelli, S.M.; Ren, X.; Vekaria, H.J.; Tripathi, P.; Zhang, L.; Zhi, W.; Bieberich, E. Novel Isolation Method Reveals Sex-Specific Composition and Neurotoxicity of Small Extracellular Vesicles in a Mouse Model of Alzheimer’s Disease. Cells 2023, 12, 1623.
- Haukedal, H.; Corsi, G.I.; Gadekar, V.P.; Doncheva, N.T.; Kedia, S.; De Haan, N.; Chandrasekaran, A.; Jensen, P.; Schiønning, P.; Vallin, S.; et al. Golgi fragmentation—One of the earliest organelle phenotypes in Alzheimer’s disease neurons. Front. Neurosci. 2023, 17, 1120086.
- Crawford, K.; Leonenko, G.; Baker, E.; Grozeva, D.; Lan-Leung, B.; Holmans, P.; Williams, J.; O’Donovan, M.C.; Escott-Price, V.; Ivanov, D.K. Golgi apparatus, endoplasmic reticulum and mitochondrial function implicated in Alzheimer’s disease through polygenic risk and RNA sequencing. Mol. Psychiatry 2023, 28, 1327–1336.
- Martinez-Curiel, R.; Jansson, L.; Tsupykov, O.; Avaliani, N.; Aretio-Medina, C.; Hidalgo, I.; Monni, E.; Bengzon, J.; Skibo, G.; Lindvall, O.; et al. Oligodendrocytes in human induced pluripotent stem cell-derived cortical grafts remyelinate adult rat and human cortical neurons. Stem Cell Rep. 2023, 18, 1643–1656.
- Hösli, L.; Zuend, M.; Bredell, G.; Zanker, H.S.; Porto De Oliveira, C.E.; Saab, A.S.; Weber, B. Direct vascular contact is a hallmark of cerebral astrocytes. Cell Rep. 2022, 39, 110599.
- Ori-McKenney, K.M.; Jan, L.Y.; Jan, Y.-N. Golgi Outposts Shape Dendrite Morphology by Functioning as Sites of Acentrosomal Microtubule Nucleation in Neurons. Neuron 2012, 76, 921–930.
- Oddoux, S.; Zaal, K.J.; Tate, V.; Kenea, A.; Nandkeolyar, S.A.; Reid, E.; Liu, W.; Ralston, E. Microtubules that form the stationary lattice of muscle fibers are dynamic and nucleated at Golgi elements. J. Cell Biol. 2013, 203, 205–213.
- Wang, J.; Fourriere, L.; Gleeson, P.A. Local Secretory Trafficking Pathways in Neurons and the Role of Dendritic Golgi Outposts in Different Cell Models. Front. Mol. Neurosci. 2020, 13, 597391.
- Jamieson, J.D.; Palade, G.E. Synthesis, intracellular transport, and discharge of secretory proteins in stimulated pancreatic exocrine cells. J. Cell Biol. 1971, 50, 135–158.
- Stephens, D.J.; Lin-Marq, N.; Pagano, A.; Pepperkok, R.; Paccaud, J.-P. COPI-coated ER-to-Golgi transport complexes segregate from COPII in close proximity to ER exit sites. J. Cell Sci. 2000, 113, 2177–2185.
- Wang, T.; Hanus, C.; Cui, T.; Helton, T.; Bourne, J.; Watson, D.; Harris, K.M.; Ehlers, M.D. Local Zones of Endoplasmic Reticulum Complexity Confine Cargo in Neuronal Dendrites. Cell 2012, 148, 309–321.
- Mikhaylova, M.; Bera, S.; Kobler, O.; Frischknecht, R.; Kreutz, M.R. A Dendritic Golgi Satellite between ERGIC and Retromer. Cell Rep. 2016, 14, 189–199.
- Stoorvogel, W.; Oorschot, V.; Geuze, H.J. A novel class of clathrin-coated vesicles budding from endosomes. J. Cell Biol. 1996, 132, 21–33.
- Kemal, S.; Richardson, H.S.; Dyne, E.D.; Fu, M. ER and Golgi trafficking in axons, dendrites, and glial processes. Curr. Opin. Cell Biol. 2022, 78, 102119.
- Teixeira, A.I.; Ilkhanizadeh, S.; Wigenius, J.A.; Duckworth, J.K.; Inganäs, O.; Hermanson, O. The promotion of neuronal maturation on soft substrates. Biomaterials 2009, 30, 4567–4572.
- Watson, P.; Townley, A.K.; Koka, P.; Palmer, K.J.; Stephens, D.J. Sec16 Defines Endoplasmic Reticulum Exit Sites and is Required for Secretory Cargo Export in Mammalian Cells. Traffic 2006, 7, 1678–1687.
- Tang, B.L.; Ong, Y.S.; Huang, B.; Wei, S.; Wong, E.T.; Qi, R.; Horstmann, H.; Hong, W. A Membrane Protein Enriched in Endoplasmic Reticulum Exit Sites Interacts with COPII. J. Biol. Chem. 2001, 276, 40008–40017.
- Lee, T.; Winter, C.; Marticke, S.S.; Lee, A.; Luo, L. Essential Roles of Drosophila RhoA in the Regulation of Neuroblast Proliferation and Dendritic but Not Axonal Morphogenesis. Neuron 2000, 25, 307–316.
- Harada, A.; Teng, J.; Takei, Y.; Oguchi, K.; Hirokawa, N. MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J. Cell Biol. 2002, 158, 541–549.
- Prigozhina, N.L.; Waterman-Storer, C.M. Protein Kinase D-Mediated Anterograde Membrane Trafficking Is Required for Fibroblast Motility. Curr. Biol. 2004, 14, 88–98.
- Cole, N.B.; Sciaky, N.; Marotta, A.; Song, J.; Lippincott-Schwartz, J. Golgi dispersal during microtubule disruption: Regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol. Biol. Cell 1996, 7, 631–650.
- Thayer, D.A.; Jan, Y.N.; Jan, L.Y. Increased neuronal activity fragments the Golgi complex. Proc. Natl. Acad. Sci. USA 2013, 110, 1482–1487.
- Salvarezza, S.B.; Deborde, S.; Schreiner, R.; Campagne, F.; Kessels, M.M.; Qualmann, B.; Caceres, A.; Kreitzer, G.; Rodriguez-Boulan, E. LIM Kinase 1 and Cofilin Regulate Actin Filament Population Required for Dynamin-dependent Apical Carrier Fission from the Trans -Golgi Network. Mol. Biol. Cell 2009, 20, 438–451.
- Bossard, C.; Bresson, D.; Polishchuk, R.S.; Malhotra, V. Dimeric PKD regulates membrane fission to form transport carriers at the TGN. J. Cell Biol. 2007, 179, 1123–1131.
- Horton, A.C.; Rácz, B.; Monson, E.E.; Lin, A.L.; Weinberg, R.J.; Ehlers, M.D. Polarized Secretory Trafficking Directs Cargo for Asymmetric Dendrite Growth and Morphogenesis. Neuron 2005, 48, 757–771.
- Corda, D.; Colanzi, A.; Luini, A. The multiple activities of CtBP/BARS proteins: The Golgi view. Trends Cell Biol. 2006, 16, 167–173.
- Bonazzi, M.; Spanò, S.; Turacchio, G.; Cericola, C.; Valente, C.; Colanzi, A.; Kweon, H.S.; Hsu, V.W.; Polishchuck, E.V.; Polishchuck, R.S.; et al. CtBP3/BARS drives membrane fission in dynamin-independent transport pathways. Nat. Cell Biol. 2005, 7, 570–580.
- Valente, C.; Turacchio, G.; Mariggiò, S.; Pagliuso, A.; Gaibisso, R.; Di Tullio, G.; Santoro, M.; Formiggini, F.; Spanò, S.; Piccini, D.; et al. A 14-3-3γ dimer-based scaffold bridges CtBP1-S/BARS to PI(4)KIIIβ to regulate post-Golgi carrier formation. Nat. Cell Biol. 2012, 14, 343–354.
- Furusawa, T.; Moribe, H.; Kondoh, H.; Higashi, Y. Identification of CtBP1 and CtBP2 as Corepressors of Zinc Finger-Homeodomain Factor δEF1. Mol. Cell Biol. 1999, 19, 8581–8590.
- Hübler, D.; Rankovic, M.; Richter, K.; Lazarevic, V.; Altrock, W.D.; Fischer, K.-D.; Gundelfinger, E.D.; Fejtova, A. Differential Spatial Expression and Subcellular Localization of CtBP Family Members in Rodent Brain. PLoS ONE 2012, 7, e39710.
- Hildebrand, J.D.; Soriano, P. Overlapping and Unique Roles for C-Terminal Binding Protein 1 (CtBP1) and CtBP2 during Mouse Development. Mol. Cell Biol. 2002, 22, 5296–5307.
- Acosta-Baena, N.; Tejada-Moreno, J.A.; Arcos-Burgos, M.; Villegas-Lanau, C.A. CTBP1 and CTBP2 mutations underpinning neurological disorders: A systematic review. Neurogenetics 2022, 23, 231–240.
- Horton, A.C.; Ehlers, M.D. Neuronal Polarity and Trafficking. Neuron 2003, 40, 277–295.
- Collins, M.O.; Husi, H.; Yu, L.; Brandon, J.M.; Anderson, C.N.G.; Blackstock, W.P.; Choudhary, J.S.; Grant, S.G.N. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 2006, 97, 16–23.
- Chen, M.; Xu, L.; Wu, Y.; Soba, P.; Hu, C. The organization and function of the Golgi apparatus in dendrite development and neurological disorders. Genes Dis. 2023, 10, 2425–2442.
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular Motors in Neurons: Transport Mechanisms and Roles in Brain Function, Development, and Disease. Neuron 2010, 68, 610–638.
- Quassollo, G.; Wojnacki, J.; Salas, D.A.; Gastaldi, L.; Marzolo, M.P.; Conde, C.; Bisbal, M.; Couve, A.; Cáceres, A. A RhoA Signaling Pathway Regulates Dendritic Golgi Outpost Formation. Curr. Biol. 2015, 25, 971–982.
- Bisbal, M.; Conde, C.; Donoso, M.; Bollati, F.; Sesma, J.; Quiroga, S.; Díaz Añel, A.; Malhotra, V.; Marzolo, M.P.; Cáceres, A. Protein Kinase D Regulates Trafficking of Dendritic Membrane Proteins in Developing Neurons. J. Neurosci. 2008, 28, 9297–9308.
- Pagliuso, A.; Valente, C.; Giordano, L.L.; Filograna, A.; Li, G.; Circolo, D.; Turacchio, G.; Marzullo, V.M.; Mandrich, L.; Zhukovsky, M.A.; et al. Golgi membrane fission requires the CtBP1-S/BARS-induced activation of lysophosphatidic acid acyltransferase δ. Nat. Commun. 2016, 7, 12148.
- Sanders, S.J.; Sahin, M.; Hostyk, J.; Thurm, A.; Jacquemont, S.; Avillach, P.; Douard, E.; Martin, C.L.; Modi, M.E.; Moreno-De-Luca, A.; et al. A framework for the investigation of rare genetic disorders in neuropsychiatry. Nat. Med. 2019, 25, 1477–1487.
- Kaler, S.G. ATP7A-related copper transport diseases—Emerging concepts and future trends. Nat. Rev. Neurol. 2011, 7, 15–29.
- Terada, K.; Schilsky, M.L.; Miura, N.; Sugiyama, T. ATP7B (WND) protein. Int. J. Biochem. Cell Biol. 1998, 30, 1063–1067.
- Bartee, M.Y.; Lutsenko, S. Hepatic copper-transporting ATPase ATP7B: Function and inactivation at the molecular and cellular level. BioMetals 2007, 20, 627.
- Dierick, H.A.; Ambrosini, L.; Spencer, J.; Glover, T.W.; Mercer, J.F.B. Molecular Structure of the Menkes Disease Gene (ATP7A). Genomics 1995, 28, 462–469.
- Tümer, Z. An Overview and Update of ATP7A Mutations Leading to Menkes Disease and Occipital Horn Syndrome. Hum. Mutat. 2013, 34, 417–429.
- Tümer, Z.; Møller, L.B.; Horn, N. Mutation Spectrum of ATP7A, the Gene Defective in Menkes Disease. In Copper Transport and Its Disorders; Leone, A., Mercer, J.F.B., Eds.; Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 1999; Volume 448, pp. 83–95.
- Guthrie, L.M.; Soma, S.; Yuan, S.; Silva, A.; Zulkifli, M.; Snavely, T.C.; Greene, H.F.; Nunez, E.; Lynch, B.; De Ville, C.; et al. Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice. Science 2020, 368, 620–625.
- Lutsenko, S.; Barnes, N.L.; Bartee, M.Y.; Dmitriev, O.Y. Function and Regulation of Human Copper-Transporting ATPases. Physiol. Rev. 2007, 87, 1011–1046.
- Morgan, M.T.; Bourassa, D.; Harankhedkar, S.; McCallum, A.M.; Zlatic, S.A.; Calvo, J.S.; Meloni, G.; Faundez, V.; Fahrni, C.J. Ratiometric two-photon microscopy reveals attomolar copper buffering in normal and Menkes mutant cells. Proc. Natl. Acad. Sci. USA 2019, 116, 12167–12172.
- Peyrard, M.; Seroussi, E.; Sandberg-Nordqvist, A.-C.; Xie, Y.-G.; Han, F.-Y.; Fransson, I.; Collins, J.; Dunham, I.; Kost-Alimova, M.; Imreh, S.; et al. The human LARGE gene from 22q12.3-q13.1 is a new, distinct member of the glycosyltransferase gene family. Proc. Natl. Acad. Sci. USA 1999, 96, 598–603.
- Clarke, N.F.; Maugenre, S.; Vandebrouck, A.; Urtizberea, J.A.; Willer, T.; Peat, R.A.; Gray, F.; Bouchet, C.; Manya, H.; Vuillaumier-Barrot, S.; et al. Congenital muscular dystrophy type 1D (MDC1D) due to a large intragenic insertion/deletion, involving intron 10 of the LARGE gene. Eur. J. Hum. Genet. 2011, 19, 452–457.
- Longman, C. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of α-dystroglycan. Hum. Mol. Genet. 2003, 12, 2853–2861.
- Satz, J.S.; Ostendorf, A.P.; Hou, S.; Turner, A.; Kusano, H.; Lee, J.C.; Turk, R.; Nguyen, H.; Ross-Barta, S.E.; Westra, S.; et al. Distinct Functions of Glial and Neuronal Dystroglycan in the Developing and Adult Mouse Brain. J. Neurosci. 2010, 30, 14560–14572.
- Holzfeind, P.J. Skeletal, cardiac and tongue muscle pathology, defective retinal transmission, and neuronal migration defects in the Largemyd mouse defines a natural model for glycosylation-deficient muscle—Eye—Brain disorders. Hum. Mol. Genet. 2002, 11, 2673–2687.
- Jo, S.; Lee, K.-H.; Song, S.; Jung, Y.-K.; Park, C.-S. Identification and functional characterization of cereblon as a binding protein for large-conductance calcium-activated potassium channel in rat brain. J. Neurochem. 2005, 94, 1212–1224.
- Ou, X.-M.; Lemonde, S.; Jafar-Nejad, H.; Bown, C.D.; Goto, A.; Rogaeva, A.; Albert, P.R. Freud-1: A Neuronal Calcium-Regulated Repressor of the 5-HT1A Receptor Gene. J. Neurosci. 2003, 23, 7415–7425.
- Piton, A.; Michaud, J.L.; Peng, H.; Aradhya, S.; Gauthier, J.; Mottron, L.; Champagne, N.; Lafreniere, R.G.; Hamdan, F.F.; S2D team. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum. Mol. Genet. 2008, 17, 3965–3974.
- Vaillend, C.; Poirier, R.; Laroche, S. Genes, plasticity and mental retardation. Behav. Brain Res. 2008, 192, 88–105.
- Lee, D.; Lee, K.-H.; Ho, W.-K.; Lee, S.-H. Target Cell-Specific Involvement of Presynaptic Mitochondria in Post-Tetanic Potentiation at Hippocampal Mossy Fiber Synapses. J. Neurosci. 2007, 27, 13603–13613.
- Tang, Y.; Zucker, R.S. Mitochondrial Involvement in Post-Tetanic Potentiation of Synaptic Transmission. Neuron 1997, 18, 483–491.
- Paidi, R.; Nthenge-Ngumbau, D.; Singh, R.; Kankanala, T.; Mehta, H.; Mohanakumar, K. Mitochondrial Deficits Accompany Cognitive Decline Following Single Bilateral Intracerebroventricular Streptozotocin. Curr. Alzheimer Res. 2015, 12, 785–795.
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1998, 1, 366–373.
- Boda, B.; Alberi, S.; Nikonenko, I.; Node-Langlois, R.; Jourdain, P.; Moosmayer, M.; Parisi-Jourdain, L.; Muller, D. The Mental Retardation Protein PAK3 Contributes to Synapse Formation and Plasticity in Hippocampus. J. Neurosci. 2004, 24, 10816–10825.
- Shree, A.; Shukla, P.C. Intellectual Disability: Definition, classification, causes and characteristics. Learn. Community-Int. J. Educ. Soc. Dev. 2016, 7, 9.
- Hayashi, S.; Inoue, Y.; Hattori, S.; Kaneko, M.; Shioi, G.; Miyakawa, T.; Takeichi, M. Loss of X-linked Protocadherin-19 differentially affects the behavior of heterozygous female and hemizygous male mice. Sci. Rep. 2017, 7, 5801.
- Takano, K.; Liu, D.; Tarpey, P.; Gallant, E.; Lam, A.; Witham, S.; Alexov, E.; Chaubey, A.; Stevenson, R.E.; Schwartz, C.E.; et al. An X-linked channelopathy with cardiomegaly due to a CLIC2 mutation enhancing ryanodine receptor channel activity. Hum. Mol. Genet. 2012, 21, 4497–4507.
- Srivastava, S.; McMillan, R.; Willis, J.; Clark, H.; Chavan, V.; Liang, C.; Zhang, H.; Hulver, M.; Mukherjee, K. X-linked intellectual disability gene CASK regulates postnatal brain growth in a non-cell autonomous manner. Acta Neuropathol. Commun. 2016, 4, 30.
- Wu, Y.; Arai, A.C.; Rumbaugh, G.; Srivastava, A.K.; Turner, G.; Hayashi, T.; Suzuki, E.; Jiang, Y.; Zhang, L.; Rodriguez, J.; et al. Mutations in ionotropic AMPA receptor 3 alter channel properties and are associated with moderate cognitive impairment in humans. Proc. Natl. Acad. Sci. USA 2007, 104, 18163–18168.
- Seo, B.A.; Cho, T.; Lee, D.Z.; Lee, J.; Lee, B.; Kim, S.-W.; Shin, H.-S.; Kang, M.-G. LARGE, an intellectual disability-associated protein, regulates AMPA-type glutamate receptor trafficking and memory. Proc. Natl. Acad. Sci. USA 2018, 115, 7111–7116.
- Zaccaria, M.L.; Di Tommaso, F.; Brancaccio, A.; Paggi, P.; Petrucci, T.C. Dystroglycan distribution in adult mouse brain: A light and electron microscopy study. Neuroscience 2001, 104, 311–324.
- Moore, S.A.; Saito, F.; Chen, J.; Michele, D.E.; Henry, M.D.; Messing, A.; Cohn, R.D.; Ross-Barta, S.E.; Westra, S.; Williamson, R.A.; et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 2002, 418, 422–425.
- Haydon, P.G. Glia: Listening and talking to the synapse. Nat. Rev. Neurosci. 2001, 2, 185–193.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
26 Sep 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No