 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antonella Leggio | -- | 4561 | 2023-09-11 13:03:32 | | | |
| 2 | Catherine Yang | + 4 word(s) | 4565 | 2023-09-12 03:00:21 | | | | |
| 3 | Catherine Yang | Meta information modification | 4565 | 2023-09-12 03:01:39 | | |
Video Upload Options
Breast cancer represents the most common cancer type and one of the major leading causes of death in the female worldwide population. Overexpression of human epidermal growth factor (HER2), a transmembrane glycoprotein related to the epidermal growth factor receptor, results in a biologically and clinically aggressive breast cancer subtype. It is also the primary driver for tumor detection and progression and, in addition to being an important prognostic factor in women diagnosed with breast cancer, HER2 is a widely known therapeutic target for drug development. In breast cancer, the overexpression of the HER2 receptor makes it a reliable biomarker and a successful therapeutic target. Several strategies have been developed to target HER2, using various targeting molecules including monoclonal antibodies and tyrosine kinase inhibitors, antibody–drug conjugates, small molecules, and peptides.
1. HER2-Targeting Monoclonal Antibodies

2. HER2 Receptor-Targeting Peptides

| No. | Sequence |
|---|---|
| Pep 1 | RIKPRKGYTR |
| Pep 2 | RIKRTNRYTR |
| Pep 3 | RIRPTRRYTR |
| Pep 4 | RIRPRNRYTR |
| Pep 5 | RIRPRKGYTR |

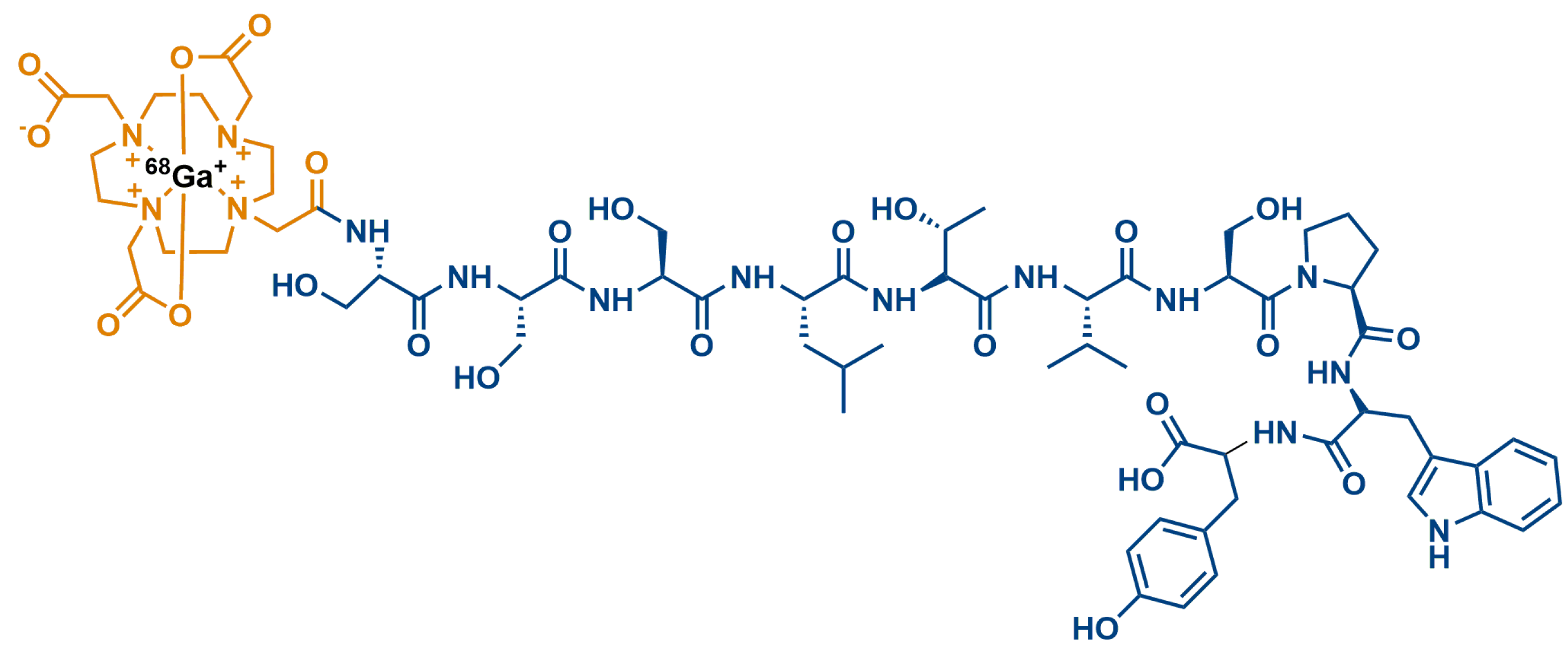
| Sequence | Name |
|---|---|
| CF-KCCYSL-NH2 | P(CC) |
| CF-KCGCYSL-NH2 | P(CGC) |
| CF-KCGGCYSL-NH2 | P(CGCG) |
| CF-KC(Acm)C(Acm)YSL-NH2 | P(C(Acm)C(Acm)) |
| CF-KCSYSL-NH2 | P(CS) |
| CF-KSCYSL-NH2 | P(SC) |
| CF-KSSYSL-NH2 | P(SS) |
| CF-KAAYSL-NH2 | P(AA) |
| CF-GYYNPT-NH2 | P(YY) |
| CF-KAAYSLGYYNPT-NH2 | cP(AA)_P(YY) |
| CF-KSCYSLGYYNPT-NH2 | cP(SC)_P(YY) |
| CF-YSLGYYNPT-NH2 | P(short)_PYY |
| CF-TAKLYPGYANYS-NH2 | scr_P(AA_YY) |
| CF-GYYNPTKAAYSL-NH2 | cP(YY)_P(AA) |
| H-KAAYSLGYYNPT-NH2 | Unlabeled cP(AA)_P(YY) |
| H-KSCYSLGYYNPT-NH2 | Unlabeled cP(SC)_P(YY) |
References
- Ferraro, E.; Drago, J.Z.; Modi, S. Implementing antibody-drug conjugates (ADCs) in HER2-positive breast cancer: State of the art and future directions. Breast Cancer Res. 2021, 23, 84.
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93.
- Goldenberg, M.M. Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clin. Ther. 1999, 21, 309–318.
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760.
- Greenblatt, K.; Khaddour, K. Trastuzumab. StatPearls: Treasure Island (FL). 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532246/ (accessed on 1 May 2023).
- Spector, N.L.; Blackwell, K.L. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2-positive breast cancer. J. Clin. Oncol. 2009, 27, 5838–5847.
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792.
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726.
- Hudis, C.A. Trastuzumab--mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51.
- Kute, T.; Stehle, J.R., Jr.; Ornelles, D.; Walker, N.; Delbono, O.; Vaughn, J.P. Understanding key assay parameters that affect measurements of trastuzumab-mediated ADCC against HER2 positive breast cancer cells. Oncoimmunology 2012, 1, 810–821.
- Arnould, L.; Gelly, M.; Penault-Llorca, F.; Benoit, L.; Bonnetain, F.; Migeon, C.; Cabaret, V.; Fermeaux, V.; Bertheau, P.; Garnier, J.; et al. Trastuzumab-based treatment of HER2-positive breast cancer: An antibody-dependent cellular cytotoxicity mechanism? Br. J. Cancer 2006, 94, 259–267.
- Gajria, D.; Chandarlapaty, S. HER2-amplified breast cancer: Mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev. Anticancer Ther. 2011, 11, 263–275.
- Cobleigh, M.A.; Vogel, C.L.; Tripathy, D.; Robert, N.J.; Scholl, S.; Fehrenbacher, L.; Wolter, J.M.; Paton, V.; Shak, S.; Lieberman, G.; et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 1999, 17, 2639–2648.
- Seidman, A.D.; Fornier, M.N.; Esteva, F.J.; Tan, L.; Kaptain, S.; Bach, A.; Panageas, K.S.; Arroyo, C.; Valero, V.; Currie, V.; et al. Weekly trastuzumab and paclitaxel therapy for metastatic breast cancer with analysis of efficacy by HER2 immunophenotype and gene amplification. J. Clin. Oncol. 2001, 19, 2587–2595.
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62.
- Rexer, B.N.; Arteaga, C.L. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: Mechanisms and clinical implications. Crit. Rev. Oncog. 2012, 17, 1–16.
- Menyhart, O.; Santarpia, L.; Gyorffy, B. A comprehensive outline of trastuzumab resistance biomarkers in HER2 overexpressing breast cancer. Curr. Cancer Drug Targets 2015, 15, 665–683.
- Slamon, D.; Pegram, M. Rationale for trastuzumab (Herceptin) in adjuvant breast cancer trials. Semin. Oncol. 2001, 28, 13–19.
- Ballantyne, A.; Dhillon, S. Trastuzumab emtansine: First global approval. Drugs. 2013, 73, 755–765.
- Eiger, D.; Pondè, N.F.; De Azambujia, E. Pertuzumab in HER2-positive early breast cancer: Current use and perspectives. Future Oncol. 2019, 15, 1823–1843.
- Amiri-Kordestani, L.; Blumenthal, G.M.; Xu, Q.C.; Zhang, L.; Tang, S.W.; Ha, L.; Weinberg, W.C.; Chi, B.; Candau-Chacon, R.; Hughes, P.; et al. FDA Approval: Ado-Trastuzumab Emtansine for the Treatment of Patients with HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2014, 20, 4436–4441.
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290.
- Diéras, V.; Miles, D.; Verma, S.; Pegram, M.; Welslau, M.; Baselga, J.; Krop, I.E.; Blackwell, K.; Hoersch, S.; Xu, J.; et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): A descriptive analysis of final overall survival results from a randomized, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 732–742.
- Krop, I.E.; Kim, S.B.; González-Martín, A.; Lo Russo, P.M.; Ferrero, J.M.; Smitt, M.; Yu, R.; Leung, A.C.; Wildiers, H. TH3RESA study collaborators. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 689–699.
- Krop, I.E.; Kim, S.B.; Martin, A.G.; LoRusso, P.M.; Ferrero, J.M.; Badovinac-Crnjevic, T.; Hoersch, S.; Smitt, M.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): Final overall survival results from a randomized open-label phase 3 trial. Lancet Oncol. 2017, 18, 743–754.
- Narayan, P.; Osgood, C.L.; Singh, H.; Chiu, H.J.; Ricks, T.K.; Chiu Yuen Chow, E.; Qiu, J.; Song, P.; Yu, J.; Namuswe, F.; et al. FDA Approval Summary: Fam-Trastuzumab Deruxtecan-Nxki for the Treatment of Unresectable or Metastatic HER2-Positive Breast Cancer. Clin Cancer Res. 2021, 27, 4478–4485.
- Mezni, E.; Vicier, C.; Guerin, M.; Sabatier, R.; Bertucci, F.; Gonçalves, A. New Therapeutics in HER2-Positive Advanced Breast Cancer: Towards a Change in Clinical Practices? Cancers 2020, 12, 1573.
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. DESTINY-Breast01 Investigators. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621.
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Abstract PD3–06: Updated results from DESTINY-breast01, a phase 2 trial of trastuzumab deruxtecan (T-DXd) in HER2 positive metastatic breast cancer. Cancer Res. 2021, 8, PD3-06.
- Bartsch, R.; Berghoff, A.S.; Furtner, J.; Marhold, M.; Bergen, E.S.; Roider-Schur, S.; Starzer, A.M.; Forstner, H.; Rottenmanner, B.; Dieckmann, K.; et al. Trastuzumab deruxtecan in HER2-positive breast cancer with brain metastases: A single-arm, phase 2 trial. Nat. Med. 2022, 28, 1840–1847.
- Swain, S.M.; Baselga, J.; Kim, S.B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Heeson, S.; et al. CLEOPATRA Study Group. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734.
- Amiri-Kordestani, L.; Wedam, S.; Zhang, L.; Tang, S.; Tilley, A.; Ibrahim, A.; Justice, R.; Pazdur, R.; Cortazar, P. First FDA approval of neoadjuvant therapy for breast cancer: Pertuzumab for the treatment of patients with HER2-positive breast cancer. Clin. Cancer Res. 2014, 20, 5359–5364.
- Nordstrom, J.L.; Gorlatov, S.; Zhang, W.; Yang, Y.; Huang, L.; Burke, S.; Li, H.; Ciccarone, V.; Zhang, T.; Stavenhagen, J.; et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Res. 2011, 13, R123.
- Schlam, I.; Nunes, R.; Lynce, F. Profile of Margetuximab: Evidence to Date in the Targeted Treatment of Metastatic HER2-positive Breast Cancer. Onco Targets Ther. 2022, 15, 471–478.
- Bang, Y.J.; Giaccone, G.; Im, S.A.; Oh, D.Y.; Bauer, T.M.; Nordstrom, J.L.; Li, H.; Chichili, G.R.; Moore, P.A.; Hong, S.; et al. First-in-human phase 1 study of margetuximab (MGAH22), an Fc-modified chimeric monoclonal antibody, in patients with HER2-positive advanced solid tumors. Ann. Oncol. 2017, 28, 855–861.
- Rugo, H.S.; Im, S.A.; Cardoso, F.; Cortés, J.; Curigliano, G.; Musolino, A.; Pegram, M.D.; Wright, G.S.; Saura, C.; Escrivá-de-Romaní, S.; et al. SOPHIA Study Group. Efficacy of Margetuximab vs Trastuzumab in Patients With Pretreated ERBB2-Positive Advanced Breast Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 573–584.
- Verdaguer, H.; Saurí, T.; Acosta, D.A.; Guardiola, M.; Sierra, A.; Hernando, J.; Nuciforo, P.; Miquel, J.M.; Molero, C.; Peiró, S.; et al. ESMO Scale for Clinical Actionability of Molecular Targets Driving Targeted Treatment in Patients with Cholangiocarcinoma. Clin. Cancer Res. 2022, 28, 1662–1671.
- Miglietta, F.; Griguolo, G.; Bottosso, M.; Giarratano, T.; Lo Mele, M.; Fassan, M.; Cacciatore, M.; Genovesi, E.; De Bartolo, D.; Vernaci, G.; et al. HER2-low-positive breast cancer: Evolution from primary tumor to residual disease after neoadjuvant treatment. NPJ Breast Cancer 2022, 8, 66.
- De Luca, S.; Verdoliva, V.; Saviano, M. Peptide Ligands Specifically Targeting HER2 Receptor and the Role Played by a Synthetic Model System of the Receptor Extracellular Domain: Hypothesized Future Perspectives. J. Med. Chem. 2020, 63, 15333–15343.
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147.
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128.
- Eberle, A.N.; Mild, G. Receptor-mediated tumor targeting with radiopeptides. Part 1. General principles and methods. J. Recept. Signal Transduct. Res. 2009, 29, 1–37.
- Spector, N.; Xia, W.; El-Hariry, I.; Yarden, Y.; Bacus, S. HER2 therapy. Small molecule HER-2 tyrosine kinase inhibitors. Breast Cancer Res. 2007, 9, 205.
- Di Gioia, M.; Leggio, A.; Liguori, A.; Perri, F. Solid-Phase Synthesis of N-Nosyl- and N-Fmoc-N-Methyl-α-amino Acids. J. Org. Chem. 2007, 72, 3723–3728.
- Ulbrich, K.; Etrych, T.; Chytil, P.; Jelínková, M.; Ríhová, B. HPMA copolymers with pH-controlled release of doxorubicin: In vitro cytotoxicity and in vivo antitumor activity. J. Control. Release 2003, 87, 33–47.
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide-Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232.
- Geng, L.; Wang, Z.; Yang, X.; Li, D.; Lian, W.; Xiang, Z.; Wang, W.; Bu, X.; Lai, W.; Hu, Z.; et al. Structure-based Design of Peptides with High Affinity and Specificity to HER2 Positive Tumors. Theranostics 2015, 5, 1154–1165.
- Eigenbrot, C.; Ultsch, M.; Dubnovitsky, A.; Abrahmsén, L.; Härd, T. Structural basis for high-affinity HER2 receptor binding by an engineered protein. Proc. Natl. Acad. Sci. USA 2010, 107, 15039–15044.
- Ekerljung, L.; Lindborg, M.; Gedda, L.; Frejd, F.Y.; Carlsson, J.; Lennartsson, J. Dimeric HER2-specific affibody molecules inhibit proliferation of the SKBR-3 breast cancer cell line. Biochem. Biophys. Res. Commun. 2008, 377, 489–494.
- Geng, L.; Wang, Z.; Jia, X.; Han, Q.; Xiang, Z.; Li, D.; Yang, X.; Zhang, D.; Bu, X.; Wang, W.; et al. HER2 Targeting Peptides Screening and Applications in Tumor Imaging and Drug Delivery. Theranostics 2016, 6, 1261–1273.
- Zhou, J.; Zou, Y.; Cai, Y.; Chi, F.; Huang, W.; Shi, W.; Qian, H. A designed cyclic peptide based on Trastuzumab used to construct peptide-drug conjugates for its HER2-targeting ability. Bioorganic Chem. 2021, 117, 105453.
- Langella, E.; Calce, E.; Saviano, M.; De Luca, S. Structural identification of an HER2 receptor model binding pocket to optimize lead compounds: A combined experimental and computational approach. Mol. BioSyst. 2016, 12, 2159–2167.
- Calce, E.; Monfregola, L.; Sandomenico, A.; Saviano, M.; De Luca, S. Fluorescence study for selecting specific ligands toward HER2 receptor: An example of receptor fragment approach. Eur. J. Med. Chem. 2013, 61, 116–121.
- De Luca, S.; Verdoliva, V.; Saviano, M.; Fattorusso, R.; Diana, D. SPR and NMR characterization of the molecular interaction between A9 peptide and a model system of HER2 receptor: A fragment approach for selecting peptide structures specific for their target. J. Pept. Sci. 2020, 26, e3231.
- Honarvar, H.; Calce, E.; Doti, N.; Langella, E.; Orlova, A.; Buijs, J.; D’Amato, V.; Bianco, R.; Saviano, M.; Tolmachev, V.; et al. Evaluation of HER2-specific peptide ligand for its employment as radiolabeled imaging probe. Sci. Rep. 2018, 8, 2998.
- Kohno, M.; Horibe, T.; Haramoto, M.; Yano, Y.; Ohara, K.; Nakajima, O.; Matsuzaki, K.; Kawakami, K. A novel hybrid peptide targeting EGFR-expressing cancers. Eur. J. Cancer 2011, 47, 773–783.
- Kawamoto, M.; Horibe, T.; Kohno, M.; Kawakami, K. HER2-targeted hybrid peptide that blocks HER2 tyrosine kinase disintegrates cancer cell membrane and inhibits tumor growth in vivo. Mol. Cancer Ther. 2013, 12, 384–393.
- Karasseva, N.G.; Glinsky, V.V.; Chen, N.X.; Komatireddy, R.; Quinn, T.P. Identification and characterization of peptides that bind human ErbB-2 selected from a bacteriophage display library. J. Protein Chem. 2002, 21, 287–296.
- Biabani Ardakani, J.; Akhlaghi, M.; Nikkholgh, B.; Hosseinimehr, S.J. Targeting and imaging of HER2 overexpression tumor with a new peptide-based 68Ga-PET radiotracer. Bioorganic Chem. 2021, 106, 104474.
- Ge, S.; Li, J.; Yu, Y.; Chen, Z.; Yang, Y.; Zhu, L.; Sang, S.; Deng, S. Review: Radionuclide Molecular Imaging Targeting HER2 in Breast Cancer with a Focus on Molecular Probes into Clinical Trials and Small Peptides. Molecules 2021, 26, 6482.
- Chen, K.; Conti, P.S. Target-specific delivery of peptide-based probes for PET imaging. Adv. Drug Deliv. Rev. 2010, 62, 1005–1022.
- Fani, M.; Maecke, H.R.; Okarvi, S.M. Radiolabeled peptides: Valuable tools for the detection and treatment of cancer. Theranostics 2012, 2, 481–501.
- Shadidi, M.; Sioud, M. Identification of novel carrier peptides for the specific delivery of therapeutics into cancer cells. FASEB J. 2003, 17, 256–258.
- Mikulová, M.B.; Mikuš, P. Advances in Development of Radiometal Labeled Amino Acid-Based Compounds for Cancer Imaging and Diagnostics. Pharmaceuticals 2021, 14, 167.
- Khodadust, F.; Ahmadpour, S.; Aligholikhamseh, N.; Abedi, S.M.; Hosseinimehr, S.J. An improved 99mTc-HYNIC-(Ser)3-LTVSPWY peptide with EDDA/tricine as co-ligands for targeting and imaging of HER2 overexpression tumor. Eur. J. Med. Chem. 2018, 144, 767–773.
- Sabahnoo, H.; Noaparast, Z.; Abedi, S.M.; Hosseinimehr, S.J. New small 99mTc-labeled peptides for HER2 receptor imaging. Eur. J. Med. Chem. 2017, 127, 1012–1024.
- Kumar, S.R.; Gallazzi, F.A.; Ferdani, R.; Anderson, C.J.; Quinn, T.P.; Deutscher, S.L. In vitro and in vivo evaluation of ⁶⁴Cu-radiolabeled KCCYSL peptides for targeting epidermal growth factor receptor-2 in breast carcinomas. Cancer Biother. Radiopharm. 2010, 25, 693–703.
- Larimer, B.M.; Thomas, W.D.; Smith, G.P.; Deutscher, S.L. Affinity maturation of an ERBB2-targeted SPECT imaging peptide by in vivo phage display. Mol. Imaging Biol. 2014, 16, 449–458.
- Kumar, S.R.; Quinn, T.P.; Deutscher, S.L. Evaluation of an 111In-radiolabeled peptide as a targeting and imaging agent for ErbB-2 receptor expressing breast carcinomas. Clin. Cancer Res. 2007, 13, 6070–6079.
- Biri-Kovács, B.; Adorján, A.; Szabó, I.; Szeder, B.; Bősze, S.; Mező, G. Structure–Activity Relationship of HER2 Receptor Targeting Peptide and Its Derivatives in Targeted Tumor Therapy. Biomolecules 2020, 10, 183.
- Luo, X.; Chen, H.; Song, Y.; Qin, Z.; Xu, L.; He, N.; Tan, Y.; Dessie, W. Advancements, challenges and future perspectives on peptide-based drugs: Focus on antimicrobial peptides. Eur. J. Pharm. Sci. 2023, 181, 106363.
- Qian, Z.; Rhodes, C.A.; McCroskey, L.C.; Wen, J.; Appiah-Kubi, G.; Wang, D.J.; Guttridge, D.C.; Pei, D. Enhancing the cell permeability and metabolic stability of peptidyl drugs by reversible bicyclization. Angew. Chem. Int. Ed. Engl. 2017, 56, 1525–1529.
- Aguirre, T.A.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.; Alonso, M.; Brayden, D. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241.
- Pichereau, C.; Allary, C. Therapeutic peptides under the spotlight. Eur. Biopharm. 2005, 5, 88–91.
- Belsito, E.; Di Gioia, M.L.; Greco, A.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. N-Methyl-N-nosyl-β3-amino Acids. J. Org. Chem. 2007, 72, 4798–4802.
- Di Gioia, M.L.; Leggio, A.; Malagrinò, F.; Romio, E.; Siciliano, C.; Liguori, A. N-Methylated α-Amino Acids And Peptides: Synthesis And Biological Activity. Mini-Rev. Med. Chem. 2016, 16, 683–690.
- Leggio, A.; Belsito, E.L.; De Marco, R.; Liguori, A.; Perri, F.; Viscomi, M.C. An Efficient Preparation of N-Methyl-α-amino Acids from N-Nosyl-α-amino Acid Phenacyl Esters. J. Org. Chem. 2010, 75, 1386–1392.
- Zhu, Q.; Chen, Z.; Paul, P.K.; Lu, Y.; Wu, W.; Qi, J. Oral delivery of proteins and peptides: Challenges, status quo and future perspectives. Acta Pharm. Sin. B 2021, 11, 2416–2448.
- Barman, P.; Joshi, S.; Sharma, S.; Preet, S.; Sharma, S.; Saini, A. Strategic Approaches to Improvise Peptide Drugs as Next Generation Therapeutics. Int. J. Pept. Res. Ther. 2023, 29, 61.
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. Bio. Drugs 2008, 22, 315–329.
- Freire Haddad, H.; Burke, J.A.; Scott, E.A.; Ameer, G.A. Clinical relevance of pre-existing and treatment-induced anti-poly(ethylene glycol) antibodies. Regen. Eng. Transl. Med. 2021, 8, 32–42.
- Costa, A.R.; Rodrigues, M.E.; Henriques, M.; Oliveira, R.; Azeredo, J. Glycosylation: Impact, control and improvement during therapeutic protein production. Crit. Rev. Biotechnol. 2014, 34, 281–299.
- Van Regenmorte, M.H. Antigenicity and immunogenicity of synthetic peptides. Biologicals 2001, 29, 209–213.
- Sauna, Z.E. Immunogenicity of Protein-Based Therapeutics. U.S. Food and Drug Administration. 2020. Available online: https://www.fda.gov/vaccines-blood-biologics/biologics-research-projects/immunogenicity-protein-based-therapeutics (accessed on 26 August 2023).
- Bray, B.L. Large-scale manufacture of peptide therapeutics by chemical synthesis. Nat. Rev. Drug Discov. 2003, 2, 587–593.
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56.

