 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Misha Ali | -- | 2315 | 2023-08-31 15:11:05 | | | |
| 2 | Lindsay Dong | Meta information modification | 2315 | 2023-09-01 02:38:28 | | |
Video Upload Options
Laccase, one of the metalloproteins, belongs to the multicopper oxidase family. It oxidizes a wide range of substrates and generates water as a sole by-product. The engineering of laccase is important to broaden their industrial and environmental applications. The general assumption is that the low redox potential of laccases is the principal obstacle, as evidenced by their low activity towards certain substrates. Therefore, the primary goal of engineering laccases is to improve their oxidation capability, thereby increasing their redox potential. Even though some of the determinants of laccase are known, it is still not entirely clear how to enhance its redox potential. However, the laccase active site has additional characteristics that regulate the enzymes’ activity and specificity. These include the electrostatic and hydrophobic environment of the substrate binding pocket, the steric effect at the substrate binding site, and the orientation of the binding substrate with respect to the T1 site of the laccase.
1. Introduction
2. An Overview of the Laccase Structure
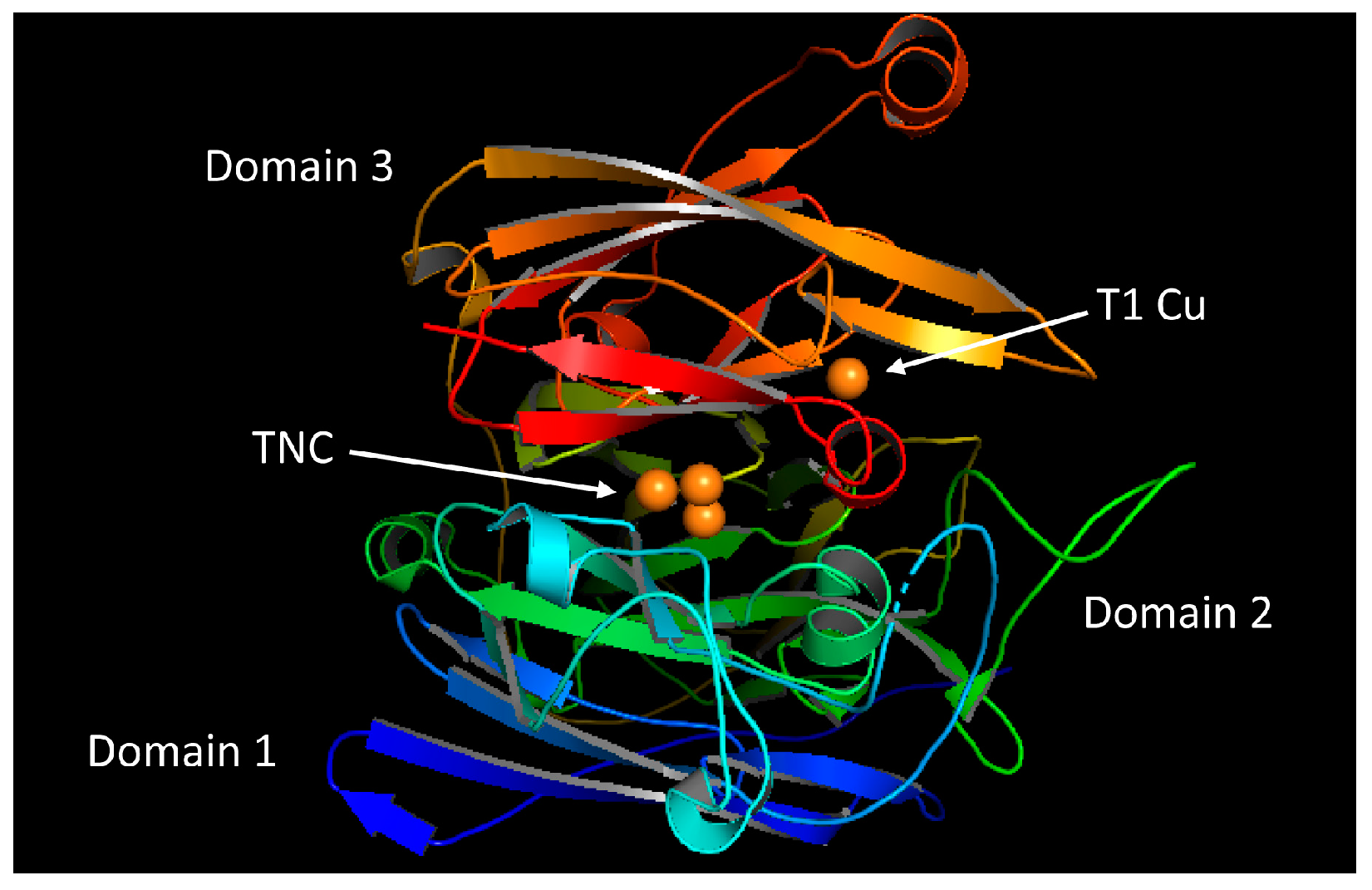
2.1. Overall Structure of the Laccase
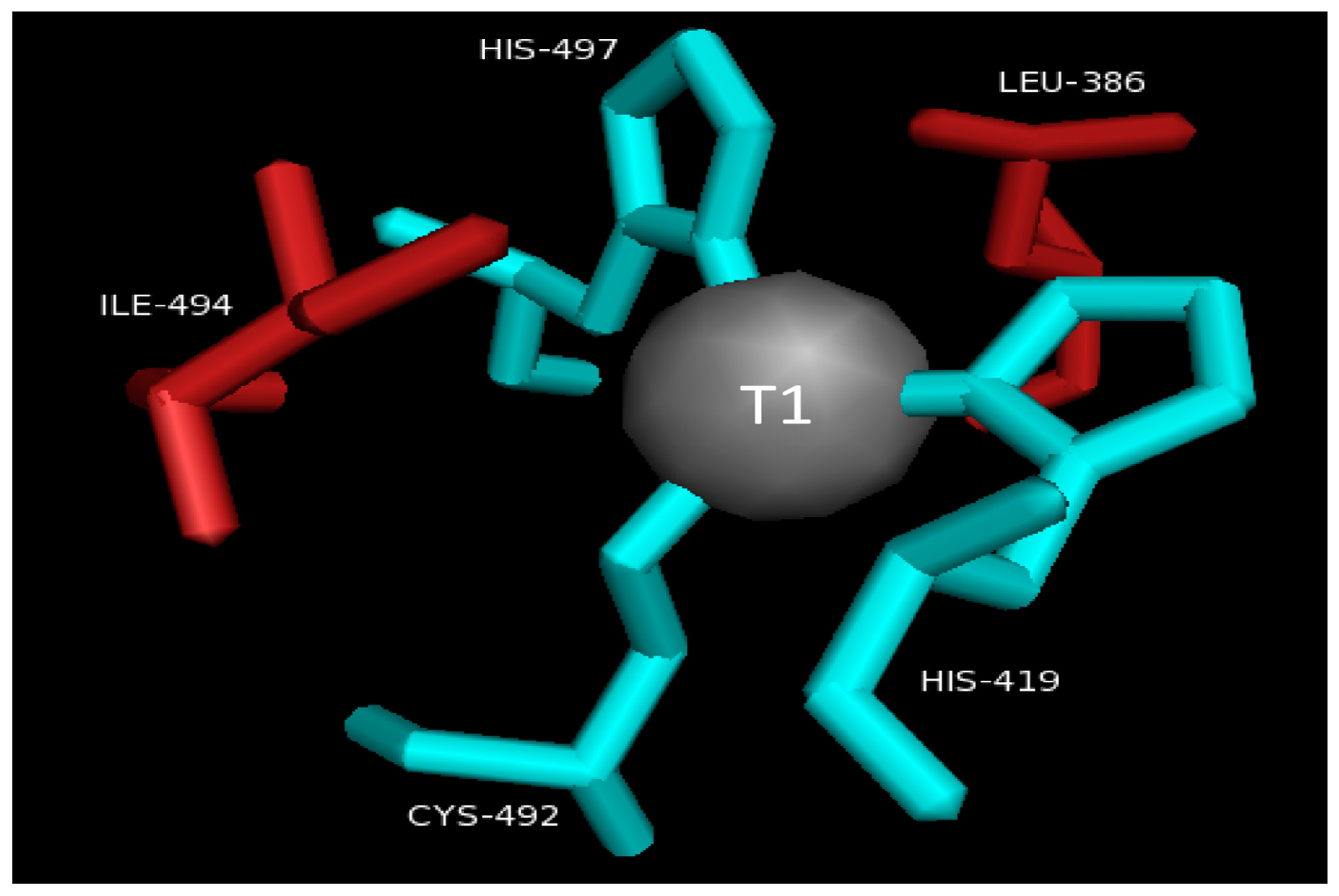
2.2. T1 Site
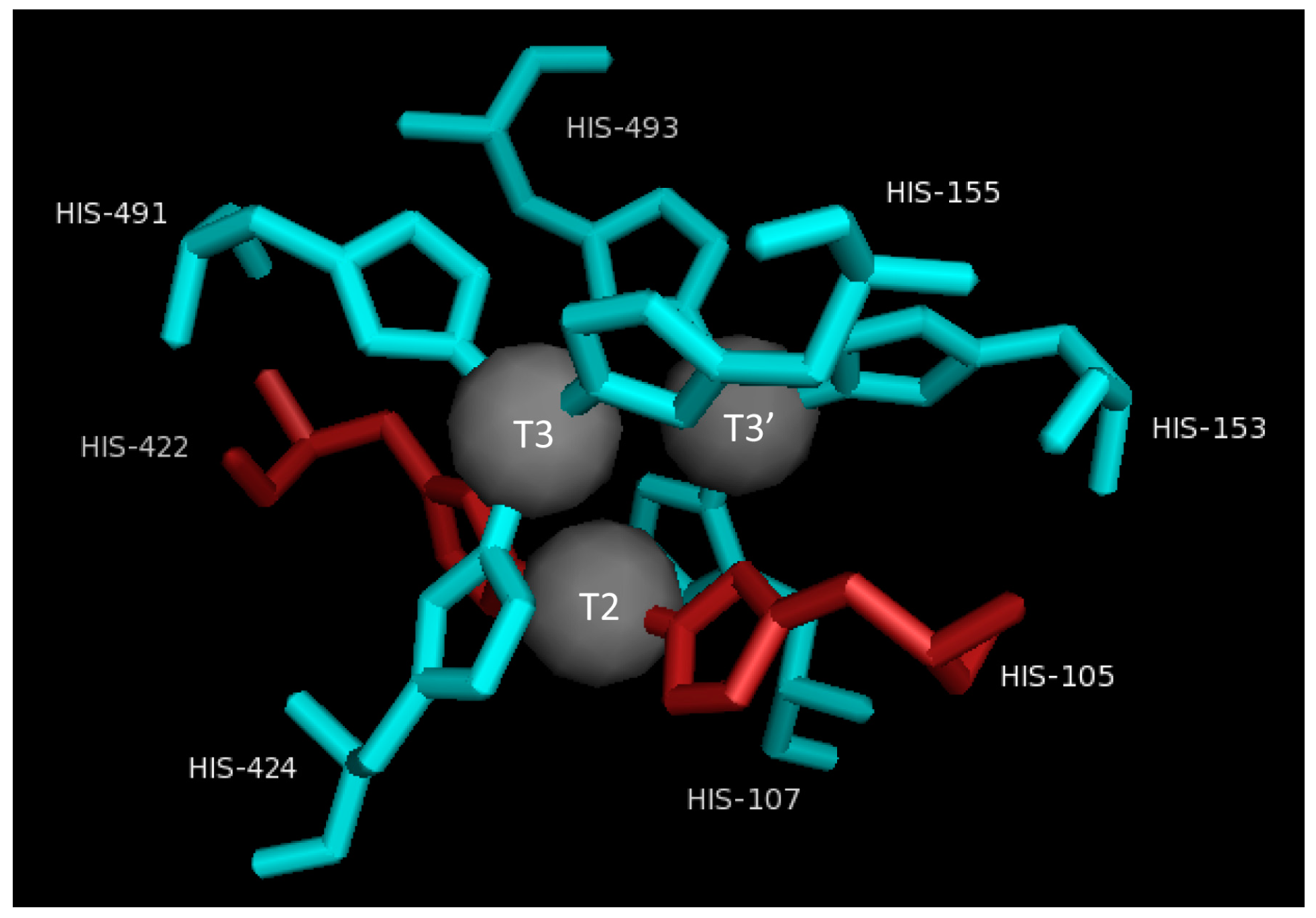
2.3. T2/T3 Site
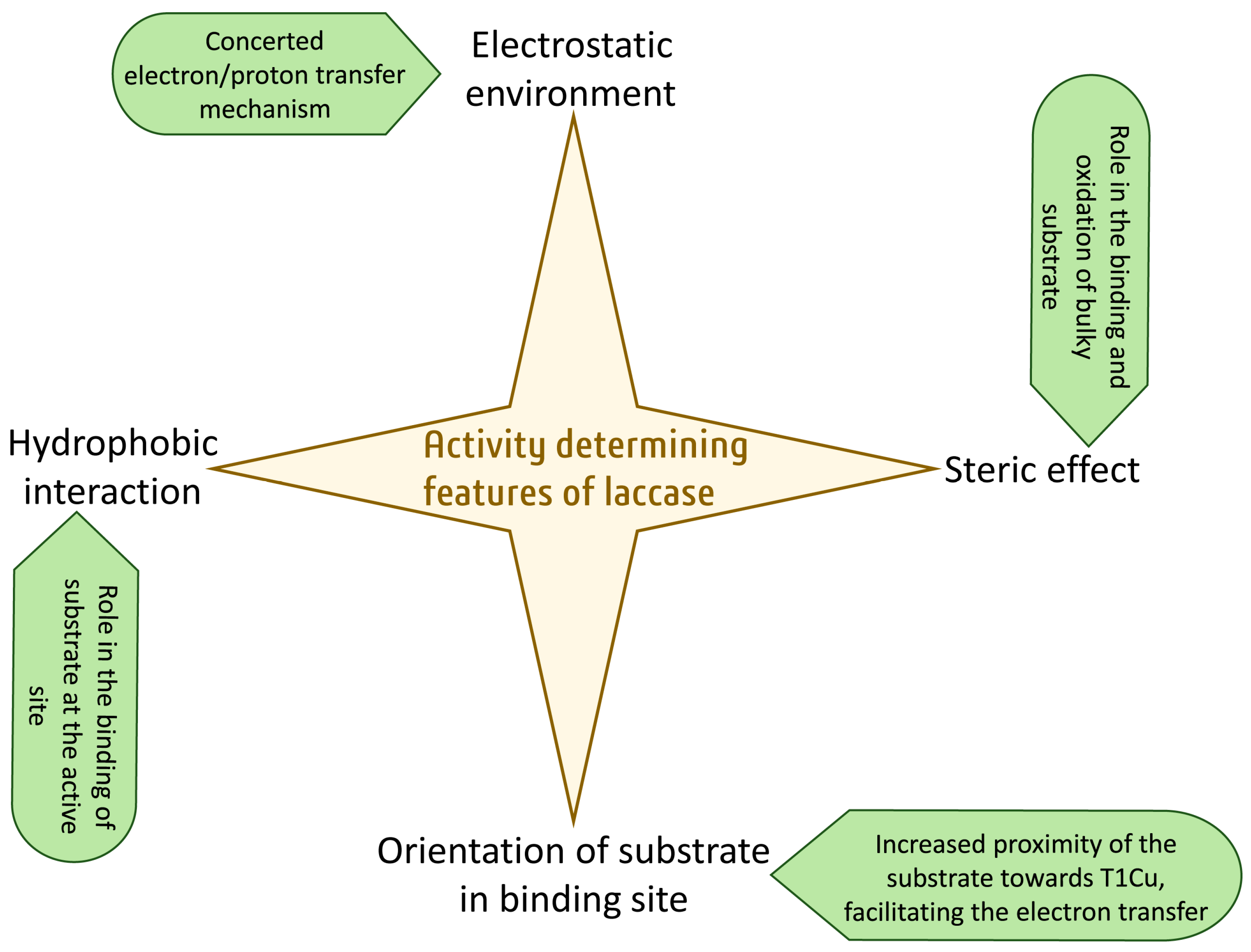
3. Characteristics That Determine Activity Other Than the Redox Potential
3.1. Electrostatic Environment of the Enzyme Pocket
3.2. Steric Hindrance Due to Bulky Structures
3.3. Orientation of Substrate in Binding Site
3.4. Hydrophobic Environment of the Enzyme Pocket
4. Conclusions
References
- Janusz, G.; Pawlik, A.; Swiderska-Burek, U.; Polak, J.; Sulej, J.; Jarosz-Wilkolazka, A.; Paszczynski, A. Laccase Properties, Physiological Functions, and Evolution. Int. J. Mol. Sci. 2020, 21, 966.
- Solomon, E.I.; Sundaram, U.M.; Machonkin, T.E. Multicopper Oxidases and Oxygenases. Chem. Rev. 1996, 96, 2563–2606.
- Kumar, A.; Ahlawat, S.; Mohan, H.; Sharma, K.K. Stabilization-destabilization and redox properties of laccases from medicinal mushroom Ganoderma lucidum and human pathogen Yersinia enterocolitica. Int. J. Biol. Macromol. 2021, 167, 369–381.
- Solomon, E.I.; Chen, P.; Metz, M.; Lee, S.K.; Palmer, A.E. Oxygen Binding, Activation, and Reduction to Water by Copper Proteins. Angew. Chem. Int. Ed. 2001, 40, 4570–4590.
- Bassanini, I.; Ferrandi, E.E.; Riva, S.; Monti, D. Biocatalysis with laccases: An updated overview. Catalysts 2020, 11, 26.
- Ayodeji, F.D.; Shava, B.; Iqbal, H.M.N.; Ashraf, S.S.; Cui, J.D.; Franco, M.; Bilal, M. Biocatalytic Versatilities and Biotechnological Prospects of Laccase for a Sustainable Industry. Catal. Lett. 2023, 153, 1932–1956.
- Cambria, M.T.; Gullotto, D.; Garavaglia, S.; Cambria, A. In silico study of structural determinants modulating the redox potential of Rigidoporus lignosus and other fungal laccases. J. Biomol. Struct. Dyn. 2012, 30, 89–101.
- Chappell, H.A.; Milliken, A.; Farmer, C.; Wendland, N.; Coward, L.; Gregory, D.J.; Johnson, C.M. Efficient remediation of 17α-ethinylestradiol by Lentinula edodes (shiitake) laccase. Biocatal. Agric. Biotechnol. 2017, 10, 64–68.
- Cardullo, N.; Muccilli, V.; Tringali, C. Laccase-mediated synthesis of bioactive natural products and their analogues. RSC Chem. Biol. 2022, 3, 614–647.
- Witayakran, S.; Ragauskas, A.J. Synthetic Applications of Laccase in Green Chemistry. Adv.Synth. Catal. 2009, 351, 1187–1209.
- Chen, Z.; Oh, W.D.; Yap, P.S. Recent advances in the utilization of immobilized laccase for the degradation of phenolic compounds in aqueous solutions: A review. Chemosphere 2022, 307, 135824.
- Mayolo-Deloisa, K.; Gonzalez-Gonzalez, M.; Rito-Palomares, M. Laccases in Food Industry: Bioprocessing, Potential Industrial and Biotechnological Applications. Front. Bioeng. Biotechnol. 2020, 8, 222.
- Stanzione, I.; Pezzella, C.; Giardina, P.; Sannia, G.; Piscitelli, A. Beyond natural laccases: Extension of their potential applications by protein engineering. Appl. Microbiol. Biotechnol. 2020, 104, 915–924.
- Backes, E.; Kato, C.G.; Corre, R.C.G.; Moreira, R.D.P.M.; Peralta, R.A.; Barros, L.; Ferreira, I.C.F.R.; Zanin, G.M.; Bracht, A.; Peralta, R.M. Laccases in food processing: Current status, bottlenecks and perspectives. Trends Food Sci. Technol. 2021, 115, 445–460.
- Minussi, R.C.; Pastore, G.M.; Duran, N. Potential applications of laccase in the food industry. Trends Food Sci. Technol. 2002, 13, 205–216.
- Hussain, A.; Bilal, M.; Rafeeq, H.; Jabeen, Z.; Afsheen, N.; Sher, F.; Kumar, V.; Bharagava, R.N.; Ferreira, L.F.R.; Iqbal, H.M. Role of laccase in the pulp and paper industry. In Nanotechnology in Paper and Wood Engineering; Elsevier: Faisalabad, Pakistan, 2022; pp. 35–60.
- Unuofin, J.O.; Falade, A.O.; Aladekoyi, O.J. Applications of microbial laccases in bioremediation of environmental pollutants: Potential issues, challenges, and prospects. Bioremediat. Environ. Sustain. 2021, 519–540.
- Leontievsky, A.A.; Vares, T.; Lankinen, P.; Shergill, J.K.; Pozdnyakova, N.N.; Myasoedova, N.M.; Kalkkinen, N.; Golovleva, L.A.; Cammack, R.; Thurston, C.F.; et al. Blue and yellow laccases of ligninolytic fungi. FEMS Microbiol. Lett. 1997, 156, 9–14.
- Koroljova-Skorobogat’ko, O.V.; Stepanova, E.V.; Gavrilova, V.P.; Morozova, O.V.; Lubimova, N.V.; Dzchafarova, A.N.; Jaropolov, A.I.; Makower, A. Purification and characterization of the constitutive form of laccase from the basidiomycete Coriolus hirsutus and effect of inducers on laccase synthesis. Biotechnol. Appl. Biochem. 1998, 28, 47–54.
- Shin, K.S.; Lee, Y.J. Purification and characterization of a new member of the laccase family from the white-rot basidiomycete Coriolus hirsutus. Arch. Biochem. Biophys. 2000, 384, 109–115.
- Jiang, Q.; Cui, Z.; Wei, R.; Nie, K.; Xu, H.; Liu, L. Feasible Cluster Model Method for Simulating the Redox Potentials of Laccase CueO and Its Variant. Front. Bioeng. Biotechnol. 2022, 10, 957694.
- Mate, D.M.; Alcalde, M. Laccase engineering: From rational design to directed evolution. Biotechnol. Adv. 2015, 33, 25–40.
- Xu, F.; Shin, W.S.; Brown, S.H.; Wahleithner, J.A.; Sundaram, U.M.; Solomon, E.I. A study of a series of recombinant fungal laccases and bilirubin oxidase that exhibit significant differences in redox potential, substrate specificity, and stability. Biochim. Biophys. Acta 1996, 1292, 303–311.
- Yaropolov, A.I.; Skorobogatko, O.V.; Vartanov, S.S.; Varfolomeyev, S.D. Laccase—Properties, Catalytic Mechanism, and Applicability. Appl. Biochem. 1994, 49, 257–280.
- Gabdulkhakov, A.; Kolyadenko, I.; Kostareva, O.; Mikhaylina, A.; Oliveira, P.; Tamagnini, P.; Lisov, A.; Tishchenko, S. Investigations of Accessibility of T2/T3 Copper Center of Two-Domain Laccase from Streptomyces griseoflavus Ac-993. Int. J. Mol. Sci. 2019, 20, 3184.
- Giardina, P.; Faraco, V.; Pezzella, C.; Piscitelli, A.; Vanhulle, S.; Sannia, G. Laccases: A never-ending story. Cell. Mol. Life Sci. 2010, 67, 369–385.
- Camarero, S.; Pardo, I.; Canas, A.I.; Molina, P.; Record, E.; Martinez, A.T.; Martinez, M.J.; Alcalde, M. Engineering platforms for directed evolution of Laccase from Pycnoporus cinnabarinus. Appl. Environ. Microbiol. 2012, 78, 1370–1384.
- Arregui, L.; Ayala, M.; Gomez-Gil, X.; Gutierrez-Soto, G.; Hernandez-Luna, C.E.; Herrera de Los Santos, M.; Levin, L.; Rojo-Dominguez, A.; Romero-Martinez, D.; Saparrat, M.C.N.; et al. Laccases: Structure, function, and potential application in water bioremediation. Microb. Cell Factories 2019, 18, 200.
- Jones, S.M.; Solomon, E.I. Electron transfer and reaction mechanism of laccases. Cell. Mol. Life Sci. 2015, 72, 869–883.
- Serrano-Posada, H.; Centeno-Leija, S.; Rojas-Trejo, S.P.; Rodríguez-Almazán, C.; Stojanoff, V.; Rudiño-Piñera, E. X-ray-induced catalytic active-site reduction of a multicopper oxidase: Structural insights into the proton-relay mechanism and O2-reduction states. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 2396–2411.
- Xu, F.; Berka, R.M.; Wahleithner, J.A.; Nelson, B.A.; Shuster, J.R.; Brown, S.H.; Palmer, A.E.; Solomon, E.I. Site-directed mutations in fungal laccase: Effect on redox potential, activity and pH profile. Biochem. J. 1998, 334 Pt 1, 63–70.
- Bertrand, T.; Jolivalt, C.; Briozzo, P.; Caminade, E.; Joly, N.; Madzak, C.; Mougin, C. Crystal structure of a four-copper laccase complexed with an arylamine: Insights into substrate recognition and correlation with kinetics. Biochemistry 2002, 41, 7325–7333.
- Autore, F.; Del Vecchio, C.; Fraternali, F.; Giardina, P.; Sannia, G.; Faraco, V. Molecular determinants of peculiar properties of a Pleurotus ostreatus laccase: Analysis by site-directed mutagenesis. Enzym. Microb. Technol. 2009, 45, 507–513.
- Loi, M.; Glazunova, O.; Fedorova, T.; Logrieco, A.F.; Mule, G. Fungal Laccases: The Forefront of Enzymes for Sustainability. J. Fungi 2021, 7, 1048.
- Rochefort, D.; Leech, D.; Bourbonnais, R. Electron transfer mediator systems for bleaching of paper pulp. Green Chem. 2004, 6, 14–24.
- Prasad, N.K.; Vindal, V.; Narayana, S.L.; Ramakrishna, V.; Kunal, S.P.; Srinivas, M. In silico analysis of Pycnoporus cinnabarinus laccase active site with toxic industrial dyes. J. Mol. Model. 2012, 18, 2013–2019.
- Miele, A.; Giardina, P.; Notomista, E.; Piscitelli, A.; Sannia, G.; Faraco, V. A semi-rational approach to engineering laccase enzymes. Mol. Biotechnol. 2010, 46, 149–156.
- Cambria, M.T.; Di Marino, D.; Falconi, M.; Garavaglia, S.; Cambria, A. Docking Simulation and Competitive Experiments Validate the Interaction Between the 2,5-Xylidine Inhibitor and Rigidoporus lignosus Laccase. J. Biomol. 2010, 27, 501–509.
- Gupta, N.; Lee, F.S.; Farinas, E.T. Laboratory evolution of laccase for substrate specificity. J. Mol. Catal. B Enzym. 2010, 62, 230–234.
- Chen, M.; Zeng, G.M.; Lai, C.; Li, J.; Xu, P.; Wu, H.P. Molecular basis of laccase bound to lignin: Insight from comparative studies on the interaction of Trametes versicolor laccase with various lignin model compounds. RSC Adv. 2015, 5, 52307–52313.
- Chen, Y.; Luo, Q.; Zhou, W.; Xie, Z.; Cai, Y.J.; Liao, X.R.; Guan, Z.B. Improving the catalytic efficiency of Bacillus pumilus CotA-laccase by site-directed mutagenesis. Appl. Microbiol. Biotechnol. 2017, 101, 1935–1944.
- Mehra, R.; Meyer, A.S.; Kepp, K.P. Molecular dynamics derived life times of active substrate binding poses explain KM of laccase mutants. RSC Adv. 2018, 8, 36915–36926.
- Kyomuhimbo, H.D.; Brink, H.G. Applications and immobilization strategies of the copper-centred laccase enzyme; a review. Heliyon 2023, 9, e13156.

