 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chao-Po Lin | -- | 5336 | 2023-08-24 18:10:51 | | | |
| 2 | Peter Tang | + 4 word(s) | 5340 | 2023-08-25 03:08:56 | | |
Video Upload Options
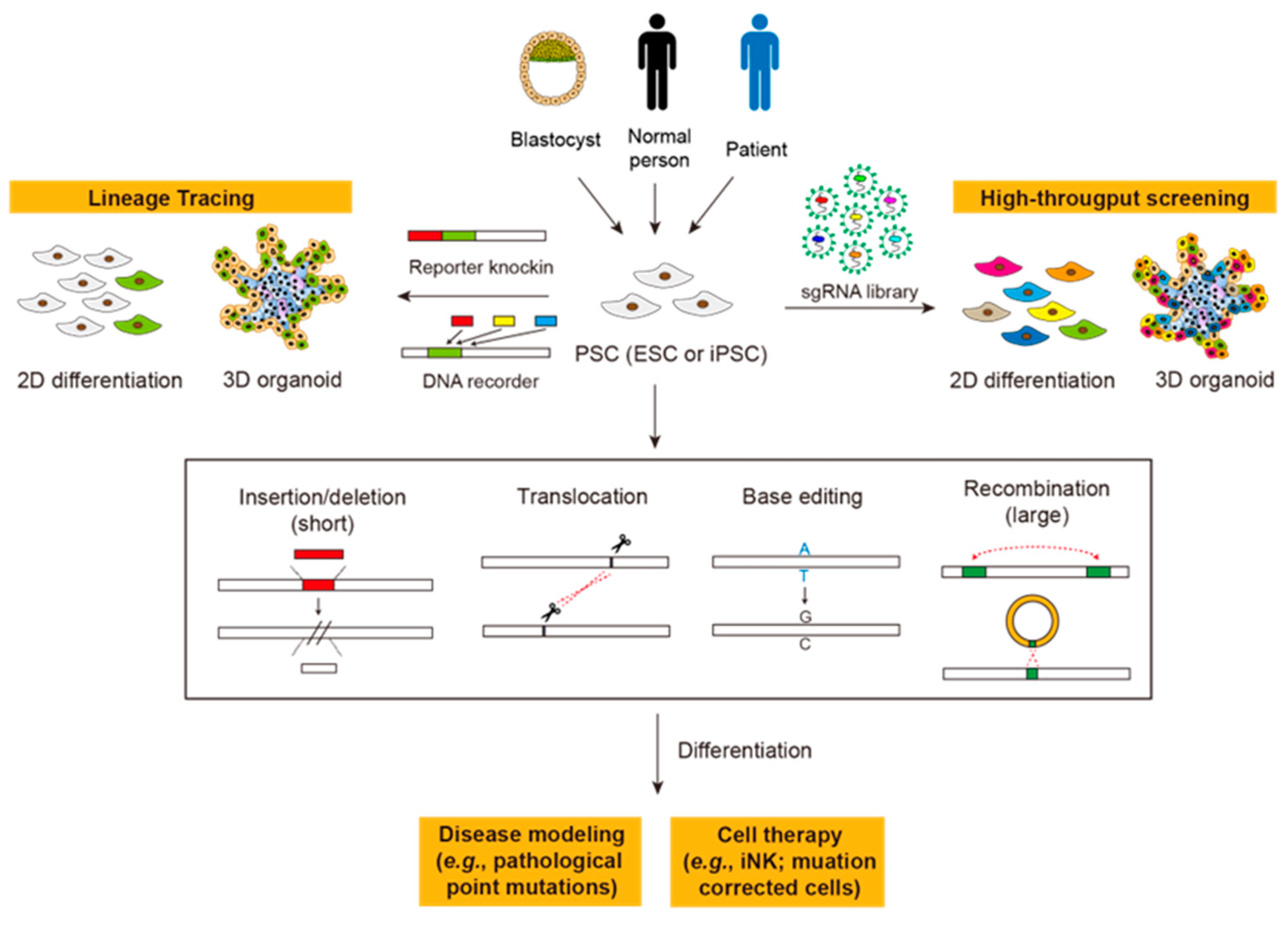
Gene editing in human pluripotent stem cells (PSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), is highly relevant to clinical cell therapy and thus should be examined with particular caution. First, since all mutations in PSCs will be carried to all their progenies, off-target edits of editors will be amplified. Second, due to the hypersensitivity of PSCs to DNA damage, double-strand breaks (DSBs) made by gene editing could lead to low editing efficiency and the enrichment of cell populations with defective genomic safeguards. In this regard, DSB-independent gene editing tools, such as base editors and prime editors, are favored due to their nature to avoid these consequences. With more understanding of the microbial world, new systems, such as Cas-related nucleases, transposons, and recombinases, are also expanding the toolbox for gene editing.
1. Introduction
|
DSB-Dependent Editor |
Base Editor |
Prime Editor |
||||||
|---|---|---|---|---|---|---|---|---|
|
ZFN |
TALEN |
SpCas9 |
CBE |
ABE |
CGBE |
AXBE/AYBE |
||
|
Type of DNA damage |
DSB |
DSB |
DSB |
SSB |
SSB |
SSB |
SSB |
SSB (PE2) or DSB (PE3) |
|
Type of editing |
Indel; Knock-in; Base mutation /correction |
Indel; Knock-in; Base mutation /correction |
Indel; Knock-in; Translocation; Base mutation /correction |
Base substitution |
Base substitution |
Base substitution |
Base substitution; |
Base substitution; Indel; Recombination |
|
p53 activation? |
Yes |
Yes |
Yes |
No |
No |
N/A |
N/A |
No |
|
On-target specificity |
+ |
+ |
++ |
++ |
+++ |
++ |
+ (C/T mix) |
+++ |
|
Off-target effects on DNA |
++ |
++ |
++ |
++ |
Very low |
++ (based on CBE) |
Very low (based on ABE) |
Low |
|
Off-target effects on RNA |
- |
- |
- |
++ |
+ |
++ (based on CBE) |
+ (based on ABE) |
- |
|
Applied in human PSCs? |
Yes |
Yes |
Yes |
Yes |
Yes |
No |
No |
Yes |
|
Clinical trial? |
Yes |
Yes |
Yes |
Yes |
Yes |
No |
No |
No |
2. DSB-Mediated Gene Editing by Programmable Nucleases
3. Base Editors
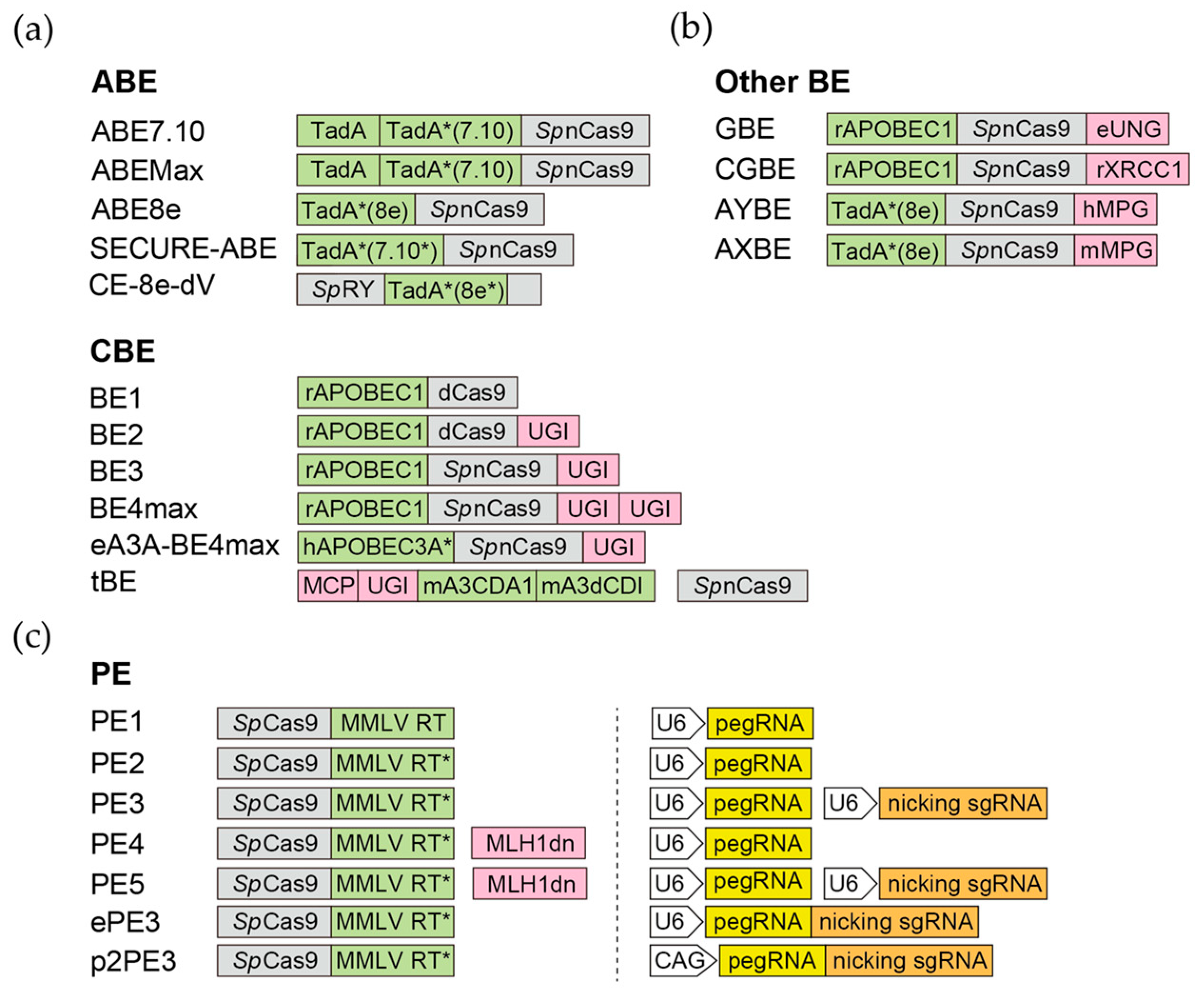
Figure 2. Base editors and prime editors. Representative versions of base editors and prime editors. The Cas9 derivatives are highlighted in grey, the key enzymatic components of BEs (a,b) and PEs (c) are highlighted in green, and the associated components are highlighted in pink. For PEs, the architectures of promoters, pegRNAs (yellow), and nicking sgRNAs (orange ) are also shown. The CAG promoter of P2PE3 can be replaced by other Pol II promoters.
3.1. Cytidine Base Editor (CBE)
3.2. Adenine Base Editor (ABE)
3.3. Glycosylase Base Editor (GBE) and C-to-G Base Editor (CGBE)
3.4. Adenine Transversion Editors (AYBE and AXBE)
3.5. Pros and Cons of Using Base Editors in PSCs
4. Prime Editor (PE)
5. New Gene Editing Tools
5.1. CRISPR-Associated Transposon (CAST)
5.2. CRISPR-Associated Serine Recombinases (twinPE and PASTE)
5.3. Retron
5.4. SeLection by Essential-Gene Exon Knock-in (SLEEK)
6. Conclusions and Future Prospects
6.1. Improving Editing Efficiencies in Human PSCs
6.2. Conditional Gene Editing
6.3. CRISPR-Cas as DNA Recorders of Cell Fates
6.4. New Systems to Be Explored
6.5. Conclusions
References
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147.
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872.
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920.
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 2011, 8, 424–429.
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412.
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362.
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801.
- Guan, J.; Wang, G.; Wang, J.; Zhang, Z.; Fu, Y.; Cheng, L.; Meng, G.; Lyu, Y.; Zhu, J.; Li, Y.; et al. Chemical reprogramming of human somatic cells to pluripotent stem cells. Nature 2022, 605, 325–331.
- Liuyang, S.; Wang, G.; Wang, Y.; He, H.; Lyu, Y.; Cheng, L.; Yang, Z.; Guan, J.; Fu, Y.; Zhu, J.; et al. Highly efficient and rapid generation of human pluripotent stem cells by chemical reprogramming. Cell Stem Cell 2023, 30, 450–459.e9.
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell 2020, 27, 523–531.
- Kim, J.Y.; Nam, Y.; Rim, Y.A.; Ju, J.H. Review of the Current Trends in Clinical Trials Involving Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2022, 18, 142–154.
- Mazzini, L.; De Marchi, F. iPSC-based research in ALS precision medicine. Cell Stem Cell 2023, 30, 748–749.
- Turan, S.; Farruggio, A.P.; Srifa, W.; Day, J.W.; Calos, M.P. Precise Correction of Disease Mutations in Induced Pluripotent Stem Cells Derived from Patients with Limb Girdle Muscular Dystrophy. Mol. Ther. 2016, 24, 685–696.
- Sumer, S.A.; Hoffmann, S.; Laue, S.; Campbell, B.; Raedecke, K.; Frajs, V.; Clauss, S.; Kaab, S.; Janssen, J.W.G.; Jauch, A.; et al. Precise Correction of Heterozygous SHOX2 Mutations in hiPSCs Derived from Patients with Atrial Fibrillation via Genome Editing and Sib Selection. Stem Cell Rep. 2020, 15, 999–1013.
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5.
- Mattapally, S.; Pawlik, K.M.; Fast, V.G.; Zumaquero, E.; Lund, F.E.; Randall, T.D.; Townes, T.M.; Zhang, J. Human Leukocyte Antigen Class I and II Knockout Human Induced Pluripotent Stem Cell-Derived Cells: Universal Donor for Cell Therapy. J. Am. Heart Assoc. 2018, 7, e010239.
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130.
- Aurora, M.; Spence, J.R. hPSC-derived lung and intestinal organoids as models of human fetal tissue. Dev. Biol. 2016, 420, 230–238.
- Frum, T.; Spence, J.R. hPSC-derived organoids: Models of human development and disease. J. Mol. Med. 2021, 99, 463–473.
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250.
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379.
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020, 15, 3380–3409.
- Bar-Ephraim, Y.E.; Kretzschmar, K.; Clevers, H. Organoids in immunological research. Nat. Rev. Immunol. 2020, 20, 279–293.
- Li, C.; Huang, J.; Yu, Y.; Wan, Z.; Chiu, M.C.; Liu, X.; Zhang, S.; Cai, J.P.; Chu, H.; Li, G.; et al. Human airway and nasal organoids reveal escalating replicative fitness of SARS-CoV-2 emerging variants. Proc. Natl. Acad. Sci. USA 2023, 120, e2300376120.
- Achberger, K.; Probst, C.; Haderspeck, J.; Bolz, S.; Rogal, J.; Chuchuy, J.; Nikolova, M.; Cora, V.; Antkowiak, L.; Haq, W.; et al. Merging organoid and organ-on-a-chip technology to generate complex multi-layer tissue models in a human retina-on-a-chip platform. eLife 2019, 8, e46188.
- Chang, C.Y.; Ting, H.C.; Liu, C.A.; Su, H.L.; Chiou, T.W.; Lin, S.Z.; Harn, H.J.; Ho, T.J. Induced Pluripotent Stem Cell (iPSC)-Based Neurodegenerative Disease Models for Phenotype Recapitulation and Drug Screening. Molecules 2020, 25, 2000.
- He, Z.; Maynard, A.; Jain, A.; Gerber, T.; Petri, R.; Lin, H.C.; Santel, M.; Ly, K.; Dupre, J.S.; Sidow, L.; et al. Lineage recording in human cerebral organoids. Nat. Methods 2022, 19, 90–99.
- Daoud, A.; Munera, J.O. Insights into Human Development and Disease from Human Pluripotent Stem Cell Derived Intestinal Organoids. Front. Med. 2019, 6, 297.
- Zhou, H.; Wang, Y.; Liu, L.P.; Li, Y.M.; Zheng, Y.W. Gene Editing in Pluripotent Stem Cells and Their Derived Organoids. Stem Cells Int. 2021, 2021, 8130828.
- Hendriks, D.; Clevers, H.; Artegiani, B. CRISPR-Cas Tools and Their Application in Genetic Engineering of Human Stem Cells and Organoids. Cell Stem Cell 2020, 27, 705–731.
- De Masi, C.; Spitalieri, P.; Murdocca, M.; Novelli, G.; Sangiuolo, F. Application of CRISPR/Cas9 to human-induced pluripotent stem cells: From gene editing to drug discovery. Hum. Genom. 2020, 14, 25.
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930.
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946.
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 2017, 545, 229–233.
- Capecchi, M.R. Altering the genome by homologous recombination. Science 1989, 244, 1288–1292.
- Koller, B.H.; Smithies, O. Inactivating the beta 2-microglobulin locus in mouse embryonic stem cells by homologous recombination. Proc. Natl. Acad. Sci. USA 1989, 86, 8932–8935.
- Rudin, N.; Sugarman, E.; Haber, J.E. Genetic and physical analysis of double-strand break repair and recombination in Saccharomyces cerevisiae. Genetics 1989, 122, 519–534.
- Rouet, P.; Smih, F.; Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 1994, 14, 8096–8106.
- Saretzki, G.; Armstrong, L.; Leake, A.; Lako, M.; von Zglinicki, T. Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells 2004, 22, 962–971.
- Maynard, S.; Swistowska, A.M.; Lee, J.W.; Liu, Y.; Liu, S.T.; Da Cruz, A.B.; Rao, M.; de Souza-Pinto, N.C.; Zeng, X.; Bohr, V.A. Human embryonic stem cells have enhanced repair of multiple forms of DNA damage. Stem Cells 2008, 26, 2266–2274.
- Fu, X.; Cui, K.; Yi, Q.; Yu, L.; Xu, Y. DNA repair mechanisms in embryonic stem cells. Cell. Mol. Life Sci. 2017, 74, 487–493.
- Tichy, E.D.; Pillai, R.; Deng, L.; Liang, L.; Tischfield, J.; Schwemberger, S.J.; Babcock, G.F.; Stambrook, P.J. Mouse embryonic stem cells, but not somatic cells, predominantly use homologous recombination to repair double-strand DNA breaks. Stem Cells Dev. 2010, 19, 1699–1711.
- Novo, N.; Romero-Tamayo, S.; Marcuello, C.; Boneta, S.; Blasco-Machin, I.; Velazquez-Campoy, A.; Villanueva, R.; Moreno-Loshuertos, R.; Lostao, A.; Medina, M.; et al. Beyond a platform protein for the degradosome assembly: The Apoptosis-Inducing Factor as an efficient nuclease involved in chromatinolysis. PNAS Nexus 2023, 2, pgac312.
- Artus, C.; Boujrad, H.; Bouharrour, A.; Brunelle, M.N.; Hoos, S.; Yuste, V.J.; Lenormand, P.; Rousselle, J.C.; Namane, A.; England, P.; et al. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010, 29, 1585–1599.
- Shivram, H.; Cress, B.F.; Knott, G.J.; Doudna, J.A. Controlling and enhancing CRISPR systems. Nat. Chem. Biol. 2021, 17, 10–19.
- Liu, G.; Lin, Q.; Jin, S.; Gao, C. The CRISPR-Cas toolbox and gene editing technologies. Mol. Cell 2022, 82, 333–347.
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236.
- Wang, J.Y.; Doudna, J.A. CRISPR technology: A decade of genome editing is only the beginning. Science 2023, 379, eadd8643.
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in cancer biology and therapy. Nat. Rev. Cancer 2022, 22, 259–279.
- Chen, L.; Hong, M.; Luan, C.; Gao, H.; Ru, G.; Guo, X.; Zhang, D.; Zhang, S.; Li, C.; Wu, J.; et al. Adenine transversion editors enable precise, efficient A*T-to-C*G base editing in mammalian cells and embryos. Nat. Biotechnol. 2023, 1–13.
- Cui, Z.; Liu, H.; Zhang, H.; Huang, Z.; Tian, R.; Li, L.; Fan, W.; Chen, Y.; Chen, L.; Zhang, S.; et al. The comparison of ZFNs, TALENs, and SpCas9 by GUIDE-seq in HPV-targeted gene therapy. Mol. Ther. Nucleic Acids 2021, 26, 1466–1478.
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471.
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Miller, B.R.; Langner, L.M.; Grunewald, J.; Joung, J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2021, 39, 41–46.
- Zhao, D.; Li, J.; Li, S.; Xin, X.; Hu, M.; Price, M.A.; Rosser, S.J.; Bi, C.; Zhang, X. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat. Biotechnol. 2021, 39, 35–40.
- Tong, H.; Wang, X.; Liu, Y.; Liu, N.; Li, Y.; Luo, J.; Ma, Q.; Wu, D.; Li, J.; Xu, C.; et al. Programmable A-to-Y base editing by fusing an adenine base editor with an N-methylpurine DNA glycosylase. Nat. Biotechnol. 2023, 1–5.
- Grunewald, J.; Zhou, R.; Lareau, C.A.; Garcia, S.P.; Iyer, S.; Miller, B.R.; Langner, L.M.; Hsu, J.Y.; Aryee, M.J.; Joung, J.K. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat. Biotechnol. 2020, 38, 861–864.
- Sakata, R.C.; Ishiguro, S.; Mori, H.; Tanaka, M.; Tatsuno, K.; Ueda, H.; Yamamoto, S.; Seki, M.; Masuyama, N.; Nishida, K.; et al. Base editors for simultaneous introduction of C-to-T and A-to-G mutations. Nat. Biotechnol. 2020, 38, 865–869.
- Xie, J.; Huang, X.; Wang, X.; Gou, S.; Liang, Y.; Chen, F.; Li, N.; Ouyang, Z.; Zhang, Q.; Ge, W.; et al. ACBE, a new base editor for simultaneous C-to-T and A-to-G substitutions in mammalian systems. BMC Biol. 2020, 18, 131.
- Zhang, X.; Zhu, B.; Chen, L.; Xie, L.; Yu, W.; Wang, Y.; Li, L.; Yin, S.; Yang, L.; Hu, H.; et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat. Biotechnol. 2020, 38, 856–860.
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424.
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016, 353, aaf8729.
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774.
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844.
- Chen, L.; Park, J.E.; Paa, P.; Rajakumar, P.D.; Prekop, H.T.; Chew, Y.T.; Manivannan, S.N.; Chew, W.L. Programmable C:G to G:C genome editing with CRISPR-Cas9-directed base excision repair proteins. Nat. Commun. 2021, 12, 1384.
- Zafra, M.P.; Schatoff, E.M.; Katti, A.; Foronda, M.; Breinig, M.; Schweitzer, A.Y.; Simon, A.; Han, T.; Goswami, S.; Montgomery, E.; et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018, 36, 888–893.
- Koblan, L.W.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; Newby, G.A.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018, 36, 843–846.
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Sola-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.J.; Liquori, A.J.; et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020, 38, 892–900.
- Liu, Y.; Zhou, C.; Huang, S.; Dang, L.; Wei, Y.; He, J.; Zhou, Y.; Mao, S.; Tao, W.; Zhang, Y.; et al. A Cas-embedding strategy for minimizing off-target effects of DNA base editors. Nat. Commun. 2020, 11, 6073.
- Rees, H.A.; Wilson, C.; Doman, J.L.; Liu, D.R. Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci. Adv. 2019, 5, eaax5717.
- Grunewald, J.; Zhou, R.; Iyer, S.; Lareau, C.A.; Garcia, S.P.; Aryee, M.J.; Joung, J.K. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 2019, 37, 1041–1048.
- Jeong, Y.K.; Yu, J.; Bae, S. Construction of non-canonical PAM-targeting adenosine base editors by restriction enzyme-free DNA cloning using CRISPR-Cas9. Sci. Rep. 2019, 9, 4939.
- Zhang, Z.; Tao, W.; Huang, S.; Sun, W.; Wang, Y.; Jiang, W.; Huang, X.; Lin, C.P. Engineering an adenine base editor in human embryonic stem cells with minimal DNA and RNA off-target activities. Mol. Ther. Nucleic Acids 2022, 29, 502–510.
- Koblan, L.W.; Arbab, M.; Shen, M.W.; Hussmann, J.A.; Anzalone, A.V.; Doman, J.L.; Newby, G.A.; Yang, D.; Mok, B.; Replogle, J.M.; et al. Efficient C*G-to-G*C base editors developed using CRISPRi screens, target-library analysis, and machine learning. Nat. Biotechnol. 2021, 39, 1414–1425.
- Chen, S.; Liu, Z.; Lai, L.; Li, Z. Efficient C-to-G Base Editing with Improved Target Compatibility Using Engineered Deaminase-nCas9 Fusions. CRISPR J. 2022, 5, 389–396.
- Chen, L.; Zhu, B.; Ru, G.; Meng, H.; Yan, Y.; Hong, M.; Zhang, D.; Luan, C.; Zhang, S.; Wu, H.; et al. Re-engineering the adenine deaminase TadA-8e for efficient and specific CRISPR-based cytosine base editing. Nat. Biotechnol. 2023, 41, 663–672.
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157.
- Chen, P.J.; Liu, D.R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 2023, 24, 161–177.
- Chen, P.J.; Hussmann, J.A.; Yan, J.; Knipping, F.; Ravisankar, P.; Chen, P.F.; Chen, C.; Nelson, J.W.; Newby, G.A.; Sahin, M.; et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 2021, 184, 5635–5652.e29.
- Adikusuma, F.; Lushington, C.; Arudkumar, J.; Godahewa, G.I.; Chey, Y.C.J.; Gierus, L.; Piltz, S.; Geiger, A.; Jain, Y.; Reti, D.; et al. Optimized nickase- and nuclease-based prime editing in human and mouse cells. Nucleic Acids Res. 2021, 49, 10785–10795.
- Jiang, T.T.; Zhang, X.O.; Weng, Z.P.; Xue, W. Deletion and replacement of long genomic sequences using prime editing. Nat. Biotechnol. 2022, 40, 227–234.
- Tao, R.; Wang, Y.H.; Hu, Y.; Jiao, Y.G.; Zhou, L.F.; Jiang, L.R.; Li, L.; He, X.Y.; Li, M.; Yu, Y.M.; et al. WT-PE: Prime editing with nuclease wild-type Cas9 enables versatile large-scale genome editing. Signal Transduct. Tar. 2022, 7, 108.
- Li, X.; Zhang, G.; Huang, S.; Liu, Y.; Tang, J.; Zhong, M.; Wang, X.; Sun, W.; Yao, Y.; Ji, Q.; et al. Development of a versatile nuclease prime editor with upgraded precision. Nat. Commun. 2023, 14, 305.
- Song, M.; Lim, J.M.; Min, S.; Oh, J.S.; Kim, D.Y.; Woo, J.S.; Nishimasu, H.; Cho, S.R.; Yoon, S.; Kim, H.H. Generation of a more efficient prime editor 2 by addition of the Rad51 DNA-binding domain. Nat. Commun. 2021, 12, 5617.
- Park, S.J.; Jeong, T.Y.; Shin, S.K.; Yoon, D.E.; Lim, S.Y.; Kim, S.P.; Choi, J.; Lee, H.; Hong, J.I.; Ahn, J.; et al. Targeted mutagenesis in mouse cells and embryos using an enhanced prime editor. Genome Biol. 2021, 22, 170.
- Velimirovic, M.; Zanetti, L.C.; Shen, M.W.; Fife, J.D.; Lin, L.; Cha, M.; Akinci, E.; Barnum, D.; Yu, T.; Sherwood, R.I. Peptide fusion improves prime editing efficiency. Nat. Commun. 2022, 13, 3512.
- Nelson, J.W.; Randolph, P.B.; Shen, S.P.; Everette, K.A.; Chen, P.J.; Anzalone, A.V.; An, M.; Newby, G.A.; Chen, J.C.; Hsu, A.; et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022, 40, 402–410.
- Chow, R.D.; Chen, J.S.; Shen, J.; Chen, S.D. A web tool for the design of prime-editing guide RNAs. Nat. Biomed. Eng. 2021, 5, 190–194.
- Hsu, J.Y.; Grunewald, J.; Szalay, R.; Shih, J.; Anzalone, A.V.; Lam, K.C.; Shen, M.W.; Petri, K.; Liu, D.R.; Joung, J.K.; et al. PrimeDesign software for rapid and simplified design of prime editing guide RNAs. Nat. Commun. 2021, 12, 1034.
- Hwang, G.H.; Jeong, Y.K.; Habib, O.; Hong, S.A.; Lim, K.; Kim, J.S.; Bae, S. PE-Designer and PE-Analyzer: Web-based design and analysis tools for CRISPR prime editing. Nucleic Acids Res. 2021, 49, W499–W504.
- Siegner, S.M.; Karasu, M.E.; Schroder, M.S.; Kontarakis, Z.; Corn, J.E. PnB Designer: A web application to design prime and base editor guide RNAs for animals and plants. BMC Bioinform. 2021, 22, 101.
- Anderson, M.V.; Haldrup, J.; Thomsen, E.A.; Wolff, J.H.; Mikkelsen, J.G. pegIT-a web-based design tool for prime editing. Nucleic Acids Res. 2021, 49, W505–W509.
- Yu, G.; Kim, H.K.; Park, J.; Kwak, H.; Cheong, Y.; Kim, D.; Kim, J.; Kim, J.; Kim, H.H. Prediction of efficiencies for diverse prime editing systems in multiple cell types. Cell 2023, 186, 2256–2272.e23.
- Li, X.; Zhou, L.; Gao, B.Q.; Li, G.; Wang, X.; Wang, Y.; Wei, J.; Han, W.; Wang, Z.; Li, J.; et al. Highly efficient prime editing by introducing same-sense mutations in pegRNA or stabilizing its structure. Nat. Commun. 2022, 13, 1669.
- Feng, Y.; Liu, S.; Mo, Q.; Liu, P.; Xiao, X.; Ma, H. Enhancing prime editing efficiency and flexibility with tethered and split pegRNAs. Protein Cell 2023, 14, 304–308.
- Liu, Y.; Yang, G.; Huang, S.; Li, X.; Wang, X.; Li, G.; Chi, T.; Chen, Y.; Huang, X.; Wang, X. Enhancing prime editing by Csy4-mediated processing of pegRNA. Cell Res. 2021, 31, 1134–1136.
- Zhang, G.; Liu, Y.; Huang, S.; Qu, S.; Cheng, D.; Yao, Y.; Ji, Q.; Wang, X.; Huang, X.; Liu, J. Enhancement of prime editing via xrRNA motif-joined pegRNA. Nat. Commun. 2022, 13, 1856.
- Huang, S.; Zhang, Z.; Tao, W.; Liu, Y.; Li, X.; Wang, X.; Harati, J.; Wang, P.Y.; Huang, X.; Lin, C.P. Broadening prime editing toolkits using RNA-Pol-II-driven engineered pegRNA. Mol. Ther. 2022, 30, 2923–2932.
- Habib, O.; Habib, G.; Hwang, G.H.; Bae, S. Comprehensive analysis of prime editing outcomes in human embryonic stem cells. Nucleic Acids Res. 2022, 50, 1187–1197.
- Zhou, M.; Tang, S.; Duan, N.; Xie, M.; Li, Z.; Feng, M.; Wu, L.; Hu, Z.; Liang, D. Targeted-Deletion of a Tiny Sequence via Prime Editing to Restore SMN Expression. Int. J. Mol. Sci. 2022, 23, 7941.
- Li, H.; Busquets, O.; Verma, Y.; Syed, K.M.; Kutnowski, N.; Pangilinan, G.R.; Gilbert, L.A.; Bateup, H.S.; Rio, D.C.; Hockemeyer, D.; et al. Highly efficient generation of isogenic pluripotent stem cell models using prime editing. Elife 2022, 11, e79208.
- Peters, J.E.; Makarova, K.S.; Shmakov, S.; Koonin, E.V. Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc. Natl. Acad. Sci. USA 2017, 114, E7358–E7366.
- Chavez, M.; Qi, L.S. Site-Programmable Transposition: Shifting the Paradigm for CRISPR-Cas Systems. Mol. Cell 2019, 75, 206–208.
- Strecker, J.; Ladha, A.; Gardner, Z.; Schmid-Burgk, J.L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. RNA-guided DNA insertion with CRISPR-associated transposases. Science 2019, 365, 48–53.
- Tou, C.J.; Orr, B.; Kleinstiver, B.P. Precise cut-and-paste DNA insertion using engineered type V-K CRISPR-associated transposases. Nat. Biotechnol. 2023, 41, 968–979.
- Lampe, G.D.; King, R.T.; Halpin-Healy, T.S.; Klompe, S.E.; Hogan, M.I.; Vo, P.L.H.; Tang, S.; Chavez, A.; Sternberg, S.H. Targeted DNA integration in human cells without double-strand breaks using CRISPR-associated transposases. Nat. Biotechnol. 2023, 1–12.
- Durrant, M.G.; Fanton, A.; Tycko, J.; Hinks, M.; Chandrasekaran, S.S.; Perry, N.T.; Schaepe, J.; Du, P.P.; Lotfy, P.; Bassik, M.C.; et al. Systematic discovery of recombinases for efficient integration of large DNA sequences into the human genome. Nat. Biotechnol. 2023, 41, 488–499.
- Anzalone, A.V.; Gao, X.D.; Podracky, C.J.; Nelson, A.T.; Koblan, L.W.; Raguram, A.; Levy, J.M.; Mercer, J.A.M.; Liu, D.R. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 2022, 40, 731–740.
- Yarnall, M.T.N.; Ioannidi, E.I.; Schmitt-Ulms, C.; Krajeski, R.N.; Lim, J.; Villiger, L.; Zhou, W.; Jiang, K.; Garushyants, S.K.; Roberts, N.; et al. Drag-and-drop genome insertion of large sequences without double-strand DNA cleavage using CRISPR-directed integrases. Nat. Biotechnol. 2023, 41, 500–512.
- Lampson, B.C.; Sun, J.; Hsu, M.Y.; Vallejo-Ramirez, J.; Inouye, S.; Inouye, M. Reverse transcriptase in a clinical strain of Escherichia coli: Production of branched RNA-linked msDNA. Science 1989, 243, 1033–1038.
- Hsu, M.Y.; Inouye, M.; Inouye, S. Retron for the 67-base multicopy single-stranded DNA from Escherichia coli: A potential transposable element encoding both reverse transcriptase and Dam methylase functions. Proc. Natl. Acad. Sci. USA 1990, 87, 9454–9458.
- Hsu, M.Y.; Eagle, S.G.; Inouye, M.; Inouye, S. Cell-free synthesis of the branched RNA-linked msDNA from retron-Ec67 of Escherichia coli. J. Biol. Chem. 1992, 267, 13823–13829.
- Linquist, S.; Cottenie, K.; Elliott, T.A.; Saylor, B.; Kremer, S.C.; Gregory, T.R. Applying ecological models to communities of genetic elements: The case of neutral theory. Mol. Ecol. 2015, 24, 3232–3242.
- Sharon, E.; Chen, S.A.; Khosla, N.M.; Smith, J.D.; Pritchard, J.K.; Fraser, H.B. Functional Genetic Variants Revealed by Massively Parallel Precise Genome Editing. Cell 2018, 175, 544–557.e16.
- Lopez, S.C.; Crawford, K.D.; Lear, S.K.; Bhattarai-Kline, S.; Shipman, S.L. Precise genome editing across kingdoms of life using retron-derived DNA. Nat. Chem. Biol. 2022, 18, 199–206.
- Kong, X.; Wang, Z.; Zhang, R.; Wang, X.; Zhou, Y.; Shi, L.; Yang, H. Precise genome editing without exogenous donor DNA via retron editing system in human cells. Protein Cell 2021, 12, 899–902.
- Allen, A.G.; Khan, S.Q.; Margulies, C.M.; Viswanathan, R.; Lele, S.; Blaha, L.; Scott, S.N.; Izzo, K.M.; Gerew, A.; Pattali, R.; et al. A highly efficient transgene knock-in technology in clinically relevant cell types. Nat. Biotechnol. 2023, 1–12.
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826.
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 2014, 3, e04766.
- Li, M.; Zhong, A.; Wu, Y.; Sidharta, M.; Beaury, M.; Zhao, X.; Studer, L.; Zhou, T. Transient inhibition of p53 enhances prime editing and cytosine base-editing efficiencies in human pluripotent stem cells. Nat. Commun. 2022, 13, 6354.
- Zhang, Z.; Baxter, A.E.; Ren, D.; Qin, K.; Chen, Z.; Collins, S.M.; Huang, H.; Komar, C.A.; Bailer, P.F.; Parker, J.B.; et al. Efficient engineering of human and mouse primary cells using peptide-assisted genome editing. Nat. Biotechnol. 2023, 1–11.
- Foss, D.V.; Muldoon, J.J.; Nguyen, D.N.; Carr, D.; Sahu, S.U.; Hunsinger, J.M.; Wyman, S.K.; Krishnappa, N.; Mendonsa, R.; Schanzer, E.V.; et al. Peptide-mediated delivery of CRISPR enzymes for the efficient editing of primary human lymphocytes. Nat. Biomed. Eng. 2023, 7, 647–660.
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015, 16, 142–147.
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548.
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542.
- Robert, F.; Barbeau, M.; Ethier, S.; Dostie, J.; Pelletier, J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015, 7, 93.
- Shin, H.R.; See, J.E.; Kweon, J.; Kim, H.S.; Sung, G.J.; Park, S.; Jang, A.H.; Jang, G.; Choi, K.C.; Kim, I.; et al. Small-molecule inhibitors of histone deacetylase improve CRISPR-based adenine base editing. Nucleic Acids Res. 2021, 49, 2390–2399.
- Perli, S.D.; Cui, C.H.; Lu, T.K. Continuous genetic recording with self-targeting CRISPR-Cas in human cells. Science 2016, 353, aag0511.
- Tang, W.; Liu, D.R. Rewritable multi-event analog recording in bacterial and mammalian cells. Science 2018, 360, aap8992.
- McKenna, A.; Findlay, G.M.; Gagnon, J.A.; Horwitz, M.S.; Schier, A.F.; Shendure, J. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 2016, 353, aaf7907.
- Choi, J.; Chen, W.; Minkina, A.; Chardon, F.M.; Suiter, C.C.; Regalado, S.G.; Domcke, S.; Hamazaki, N.; Lee, C.; Martin, B.; et al. A time-resolved, multi-symbol molecular recorder via sequential genome editing. Nature 2022, 608, 98–107.
- Pausch, P.; Al-Shayeb, B.; Bisom-Rapp, E.; Tsuchida, C.A.; Li, Z.; Cress, B.F.; Knott, G.J.; Jacobsen, S.E.; Banfield, J.F.; Doudna, J.A. CRISPR-CasPhi from huge phages is a hypercompact genome editor. Science 2020, 369, 333–337.
- Li, Z.; Zhong, Z.; Wu, Z.; Pausch, P.; Al-Shayeb, B.; Amerasekera, J.; Doudna, J.A.; Jacobsen, S.E. Genome editing in plants using the compact editor CasPhi. Proc. Natl. Acad. Sci. USA 2023, 120, e2216822120.
- Al-Shayeb, B.; Skopintsev, P.; Soczek, K.M.; Stahl, E.C.; Li, Z.; Groover, E.; Smock, D.; Eggers, A.R.; Pausch, P.; Cress, B.F.; et al. Diverse virus-encoded CRISPR-Cas systems include streamlined genome editors. Cell 2022, 185, 4574–4586.e16.
- Karvelis, T.; Druteika, G.; Bigelyte, G.; Budre, K.; Zedaveinyte, R.; Silanskas, A.; Kazlauskas, D.; Venclovas, C.; Siksnys, V. Transposon-associated TnpB is a programmable RNA-guided DNA endonuclease. Nature 2021, 599, 692–696.
- Altae-Tran, H.; Kannan, S.; Demircioglu, F.E.; Oshiro, R.; Nety, S.P.; McKay, L.J.; Dlakic, M.; Inskeep, W.P.; Makarova, K.S.; Macrae, R.K.; et al. The widespread IS200/IS605 transposon family encodes diverse programmable RNA-guided endonucleases. Science 2021, 374, 57–65.
- Sasnauskas, G.; Tamulaitiene, G.; Druteika, G.; Carabias, A.; Silanskas, A.; Kazlauskas, D.; Venclovas, C.; Montoya, G.; Karvelis, T.; Siksnys, V. TnpB structure reveals minimal functional core of Cas12 nuclease family. Nature 2023, 616, 384–389.
- Han, D.; Xiao, Q.; Wang, Y.; Zhang, H.; Dong, X.; Li, G.; Kong, X.; Wang, S.; Song, J.; Zhang, W.; et al. Development of miniature base editors using engineered IscB nickase. Nat. Methods 2023, 20, 1029–1036.
- Saito, M.; Xu, P.; Faure, G.; Maguire, S.; Kannan, S.; Altae-Tran, H.; Vo, S.; Desimone, A.; Macrae, R.K.; Zhang, F. Fanzor is a eukaryotic programmable RNA-guided endonuclease. Nature 2023, 1–3.
- Jiang, K.; Lim, J.; Sgrizzi, S.; Trinh, M.; Kayabolen, A.; Yutin, N.; Koonin, E.V.; Abudayyeh, O.O.; Gootenberg, J.S. Programmable RNA-guided endonucleases are widespread in eukaryotes and their viruses. bioRxiv 2023.
- Wilkinson, M.E.; Frangieh, C.J.; Macrae, R.K.; Zhang, F. Structure of the R2 non-LTR retrotransposon initiating target-primed reverse transcription. Science 2023, 380, 301–308.



