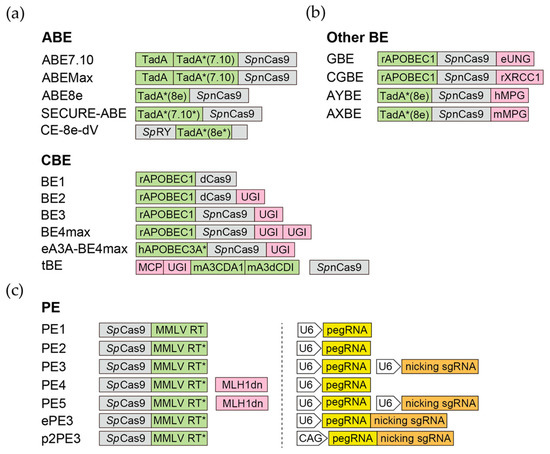
Since DSB-mediated editing raises concerns for undesired mutations in PSCs, base editors, which make base substitutions without introducing DSBs, are reasonably favored for gene editing in PSCs. Theoretically, six base editors will be needed for “any base to any base” substitutions (please refer to Chen et al. [
125] for the illustration). Yet, pathogenic point mutations in humans are not evenly distributed (also Chen et al. [
125] for the statistics), making it possible to cover most human diseases with fewer editors. The first two base editors reported, the cytidine base editor (CBE) and the adenine base editor (ABE), were realized by David Liu’s lab in 2016 and 2017, respectively [
112,
122]. CBE converts cytidines to thymines on one strand and thus can be used to create both C>T (
C•T→
T•A) and G>A (G•
C→A•
T) substitutions. Similarly, ABE substitutes adenines with guanines and thus can make both A>G (
A•T→
G•A) and T>C (T•
A→C•
G) substitutions. Those four types of editing cover ~60% of edits needed for correcting pathogenic mutations [
125]. In 2020, two labs reported the glycosylase base editor that can make C>G and G>C conversions, which constitute ~10% of pathological mutations [
123,
124]. Recently, new editors were reported to be able to convert A to C or T [
125,
126], covering another ~25% of pathological mutations. In addition to these single-base editors, dual-base editors, which fuse two types of single-base editors, were also created to introduce multiple substitutions [
127,
128,
129,
130]. Together, these base editors constitute a tool collection for introducing or correcting point mutations in somatic or stem cells.
CBEs take advantage of the cytidine deaminase activity, which converts cytidines to uracils (U), equivalent to T in base pairing, for base substitution. The rat APOBEC1 (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 1, or rAPOBEC1) and the sea lamprey AID (activation-induced cytidine deaminase) were first employed in the BE series [
131] and Target-AID [
132], respectively. Cytidine deaminases from different species, including the CDA, AID, and APOBEC3 family, are also employed to build CBEs with different editing efficiencies and sequence preferences [
133]. BE3 and BE4 are the third and fourth generation of BE, containing nCas9, rAPOBEC1, as well as one (BE3) or two (BE4) copies of uracil DNA glycosylase inhibitor (UGI) from the
Bacillus subtilis bacteriophage [
131]. UGI can suppress the repair of U by inhibiting the uracil DNA glycosylase (UNG), which excises uracil bases to form abasic sites for base excision repair (BER). To date, more than thirty CBE variants have been made by changing the editing window, expanding the PAM compatibility, or increasing/decreasing their on-target/off-target activities [
134]. Notably, despite its successful application in multiple cell lines, BE3 exhibits lower editing efficiency in human PSCs [
135] and may need further optimization for better efficiency [
136].
In 2017, David Liu’s lab reported the first ABE, which realized the A>G and T>C substitutions [
122]. Theoretically, deamination of adenosines results in inosines (I), which could complement with cytidines and be eventually converted to guanines by the mismatch repair. However, there are no known DNA adenine deaminases. To solve this issue, Gaudelli et al. subjected the
E. coli tRNA adenine deaminase TadA to extensive evolution to alter its activity towards DNA [
122]. ABE7.10, which contains a wild-type TadA and an evolved TadA* (contains 14 amino acid substitutions in the catalytic domain), was finally retrieved with the highest efficiency of converting A•T to G•C [
122]. This breakthrough opens the gate for the subsequent derivation of ABE variants with improved nuclear localization/expression (ABEmax) [
137] or smaller size and faster editing kinetics (ABE8e) [
138]. Recently, several groups, including us, modified those ABEs to further reduce their off-targeting activities [
139,
140,
141] or expand the PAM compatibility [
142,
143]. Notably, compared with CBE, ABE exhibits high product purity and low rates of indels in mammalian cells, including human PSCs, possibly due to their lack of efficient glycosylase to initiate BER [
122].
As mentioned above, the A>C substitution is required to reverse ~25% pathological point mutations. Recently, two breakthrough studies reported base editors that can transverse A to C or T in mammalian cells [
125,
126]. Both groups employ a strategy similar to GBE/CGBE, but aim at creating the AP site on A instead of C (in the GBE/CGBE case), which is expected to be mutagenized by the DNA repair pathway. To achieve this, Tong et al. constructed the adenine transverse base editor (AYBE, Y = C or T) by fusing the ABE8e with an engineered human hypoxanthine glycosylase enzyme, N-methylpurine DNA glycosylase (MPG, also called AAG). MPG excises the hypoxanthine group from the inosine produced by ABE, resulting in an AP site. The AP site will then be processed by the translesion synthesis pathway and replaced by C or T as the most common outcomes [
126]. Chen et al. employed a similar strategy but used mouse AAG (MPG) instead of the human one, creating the AXBE. Importantly, by mutagenesis and Cas embedding strategies, Chen et al. further created the ACBE-Q editor, which exhibited high A>C activity and reduced A>G bystander substitutions [
125]. Although the purity and efficiency of adenine transversion editors remain to be improved, these two editors have substantially expanded the potential of base editing.
4. Prime Editor (PE)
In 2019, a versatile gene-editing tool, prime editor (PE), was developed by the Liu lab [
166,
167]. The two essential components of the PE system are the editor, a nCas9 fused with reverse transcriptase (RT), and a single engineered prime editing guide RNA (pegRNA) which consists of the sgRNA and the intended sequence to edit. After being guided to the target site by the sgRNA component of the pegRNA, the nCas9 generates a nick in the single-stranded R-loop of the target site. The pegRNA then hybridizes with the nicked target DNA strand and serves as the template for RT to polymerize the desired sequence onto the nicked target DNA. After resolving the flap of the edited DNA by DNA repair machinery, the desired sequence will be incorporated into the genome. Decided by the templates, PE can make all types of base substitutions or insertions/deletions of small DNA fragments in mammalian cells [
166,
167].
PEs have evolved through multiple versions. The original version of prime editor (PE1) uses the wild-type RT from Moloney murine leukemia virus (MMLV) [
166]. Although PE1 can make all gene edits in human cells, the gene editing efficiency is low, typically <5% [
166]. In PE2, five mutations are introduced to MMLV-RT to enhance its thermostability, processivity, and binding affinity to the template. The gene editing efficiency of PE2 is increased 1.6- to 5.1-fold compared to PE1 in human cells [
166]. On the basis of PE2, PE3 includes an additional sgRNA to direct the nCas9 component of the prime editor to also nick the non-edited strand, promoting the replacement of the non-edited strand with the sequence complementary to the edited DNA [
166]. The gene editing efficiency of PE3 is further increased 1.5- to 4.2-fold compared to PE2 in HEK293T cells [
166]. PE4 and PE5 prime editing systems are developed by the transient expression of an engineered mismatch repair (MMR)-inhibiting protein, MLH1dn, with PE2 and PE3, respectively. The rationale behind this design is that the MMR was found to strongly antagonize prime editing and promote the generation of undesired indel byproducts [
168]. Compared with PE2 and PE3 systems, PE4 and PE5 prime editing systems enhance the editing efficiency by an average of 7.7- and 2.0-fold, respectively [
168]. PEmax, which contains R221K/N394K mutations in Cas9, two NLS (nuclear localization signal) tags, and a codon-optimized MMLV-RT, exhibits further elevated editing efficacy [
168]. Finally, replacing the nCas9 with the DSB-making Cas9 also significantly enhances the editing efficiency [
169,
170,
171,
172]. Notably, since DSBs are made by late versions of PE (since PE3), their side effects need to be determined, particularly in PSCs.
The PE system was also modified or optimized by researchers from the perspectives of editors or pegRNAs. The modification on editors is mostly by fusing with other proteins to enhance the editor’s performance. In the hyPE2 design, the Rad51 DNA-binding domain is inserted between nCas9 and RT to facilitate reverse transcription [
173]. Fusion of the chromatin-modulating peptide to PE3 (CMP-PE3) or a DNA repair-related peptide to PE2 (IN-PE2) can significantly increase the editing efficiency in mammalian cells [
174,
175]. On the other hand, the pegRNA design is optimized by various rationales. The pegRNA contains a primer binding site (PBS) sequence to trigger the reverse transcription, whose length greatly affects the editing efficiency [
176]. Several algorithms or approaches were developed to optimize the PBS length or the pegRNA sequence [
177,
178,
179,
180,
181,
182,
183]. Modifying the pegRNA by stabilizing its secondary structure or preventing its circularization also enhances the editing efficiency [
176,
183,
184,
185,
186].
The efficiencies of PEs vary widely, depending on the genomic context, the pegRNA design, and the cell type. In our work, PE editing efficiencies on the same sites are consistently lower in PSCs compared with immortalized cells, such as HEK293T [
187]. The causes of such differences remain unclear. One possibility of the low efficiency could be simply the level of PE/pegRNA expressed in cells (see Conclusion and Future Prospects). The high MMR repair capacity of PSCs could also result in this low editing efficiency, as overexpressing MLH1dn (PE4/5) enhanced the editing efficiency of PE in PSCs [
187]. Interestingly, inhibition of p53 by SV40 large T antigen (SV40LT) further increases the editing efficiency, suggesting p53 plays a role in modulating PE-mediated edits [
187]. Despite the involvement of p53, our and other researchers’ results suggest that PE-editing, in the presence or absence of editing boosters (MLH1dn or SV40LT), does not lead to off-target mutations beyond the background level [
187,
188].
Although the efficiency remains to be improved, prime editing has been successfully applied for gene editing in human PSCs to induce nucleotide substitutions or small insertions/deletions. Habib et al. used PE to correct a liver disease-related mutation of
SERPINA1 in patient-derived human iPSCs. PE was also used to precisely delete the intronic splicing silencer-N1 (ISS-N1) within survival motor neuron 2 (
SMN2) to rescue full-length SMN expression in human iPSCs derived from spinal muscular atrophy (SMA) patients [
189]. Finally, Li. et al. reported that delivering PE and pegRNA in the mRNA form greatly enhances the editing efficiency on multiple sites, which greatly facilitates applying PEs in human PSCs [
190]. However, whether this approach can generally increase the efficiencies of PEs on different target sites remains to be investigated. Finally, compared with the base editors (CBEs or ABEs), PEs exhibit lower efficiency but fewer bystander edits. Importantly, the WGS confirmed that PE does not lead to off-target mutations in the genome in PSCs [
188]. Together, these results suggest that PEs are promising editing tools to for PSCs, although their caveats remain to be solved.
5. New Gene Editing Tools
Although DSB-independent editing tools currently used are favored in PSCs, one major restriction of those tools is the inability or low efficiency to insert large DNA fragments. PEs can insert short DNA fragments (<20 nt), yet their efficiencies drop dramatically with the increased length of insertion [
187]. This property does not meet the need of many clinical applications (e.g., CAR-iNK), which call for much larger insertions. Currently, several new gene editing tools have been developed for knocking in larger DNA fragments or to enrich cell populations containing these insertions. Of note, some of these tools are still under development and need improvements for applying in mammalian cells or PSCs.
5.1. CRISPR-Associated Transposon (CAST)
As the name suggests, the CRISPR-associated transposon (CAST) is the transposon containing a specific subtype of CRISPR-Cas systems [
191]. Compared with RNA-guided endonucleases that function in the defense against MGE (mobile genetic elements), this specific CRISPR-Cas subtype is employed for RNA-guided transposition [
192]. The mechanisms of two types of CASTs, CAST I-F and CAST V-K, were elucidated in prokaryotes [
193]. As the recognition–integration process is independent of HDR, transposon-based CRISPR systems hold great expectation for inserting large DNA fragments into specific sites in eukaryotic cells. Recently, a system based on CAST, the HE-assisted large-sequence integrating CAST-complex (HELIX), has been able to insert DNA fragments into exogenous plasmids in human cells [
194]. Furthermore, Lampe et al. reported that with the help of bacterial ClpX, Type I-F CAST could reach single-digit efficiencies in human endogenous genes [
195]. Despite this success, CAST is still ineffective in editing human endogenous genes, possibly due to the unidentified regulatory factors, components, or features of eukaryotic chromatin. More understanding of structures and mechanisms of CASTs could facilitate their application in eukaryotic cells, even PSCs.
5.2. CRISPR-Associated Serine Recombinases (twinPE and PASTE)
Another strategy to integrate large DNA fragments is using recombinases, which integrate MGE into bacterial genomes on attachment sites, the specific sequences into which the payloads will be inserted. Recently, thousands of large serine recombinases (LSRs) and DNA attachment sites were predicted using computational approaches, and over 60 new LSRs were experimentally validated in human cells [
196]. In combination with TwinPE, which exhibits superior ability to insert the landing pad sequence into the desired site, large DNA fragment insertion can be mediated by the site-specific serine recombinase/integrase, Bxb1 [
197]. Another approach with a similar concept, PASTE (programmable addition via site-specific targeting elements), uses the PE-Bxb1 fusion protein and the pegRNA containing the attachment sequence (atgRNA for attachment site-containing guide RNA) [
198]. Both twinPE and PASTE can insert large DNA fragments ranging from 5.6~36 kb into human immortalized cells or cancer cells, sufficient for most purposes. It will be valuable to test the efficiencies of those tools in PSCs.
5.3. Retron
Retrons are non-transposable retroelements firstly identified in prokaryotes [
199]. A typical retron contains a reverse transcriptase (RT) and a template sequence, on which the RT acts to create the multi-copy single-stranded DNA (msDNA) [
200,
201]. The msDNAs are then joined to their template RNAs by a 2′–5′ phosphodiester bond, forming a special DNA–RNA hybrid structure [
201]. Although the functions of retrons in their hosts remain poorly understood [
202], the retron scaffold has been modified for the purpose of genome editing: a sgRNA can be added to the RNA component of the retron to guide it to the target site in the presence of Cas, while the desired donor sequence can be inserted into the retron scaffold and retro-transcribed into the msDNA [
203]. As the consequence, those msDNAs containing desired sequences will be enriched in the proximity of the sgRNA-guided cleavage site and be used as the donor template for HR [
203]. This system, named CRISPEY (Cas9 retron precise parallel editing via homology), has been employed in yeasts for massive parallel genome editing [
203]. Recently, retrons have been successfully applied for gene editing in mammalian cells, although their efficiencies remain low [
204,
205]. Other concerns for the retron system are its DSB-dependent HDR mechanism and the limited fragment size (~700 bp) that can be inserted into the retron framework. Together, more mechanistic studies are required to apply retrons in mammalian cells.
5.4. SeLection by Essential-Gene Exon Knock-in (SLEEK)
Precise knock-in of genes at desired, endogenous sites is required for many clinical purposes. However, the desired knock-in mediated by CRISPR-Cas9-induced HDR is usually mixed or even overwhelmed by undesired indels generated by NHEJ. Recently, a simple but efficient approach to enrich cells with correct knock-in was developed. In SLEEK (selection by essential-gene exon knock-in), the donor DNA fragment is targeted to a site within an exon of an essential gene. The cargo template is designed in a way that the correct knock-in will retain the essential gene function, while all the cells containing undesired products get wiped out without the need for drug selection [
206]. Importantly, this method has been applied in iPSCs to knock-in
CD16 and
mbIL-15, which enhance the anti-tumor activity and persistence of iNK cells [
206]. Although the WGS of those iPSC clones is still needed to evaluate the consequences of DSB-dependent HDR, this method provides a great advantage in saving the cost of generating clinical level PSCs.
6. Conclusions and Future Prospects
6.1. Improving Editing Efficiencies in Human PSCs
Considering the laborious process to isolate and characterize single-cell derived colonies, improving the gene editing efficiencies is crucial to apply gene editing tools in human PSCs. The Doudna and Church groups demonstrated that Cas9-mediated gene editing in PSCs is less efficient than in other somatic cells [
82,
207]. Previous studies also reported decreased editing efficiencies of CBE and PE in human PSCs [
208]. However, it is over-simplistic to directly compare the editing efficiencies between somatic/immortal cells and PSCs. Human PSCs are notoriously hard to deliver exogenous genes with high copies. Thus, the low editing efficiencies of editors in PSCs could be simply due to their low expression levels compared to cell lines that can be easily transfected. In agreement with this notion, recent studies suggested that PE delivered in the mRNA form greatly enhances the editing efficiency compared to other forms, such as plasmids or RNP complexes [
190]. Recent studies also showed that delivering Cas RNPs (Cas9 or Cas12) by cell-penetrating peptides greatly enhances editing efficiencies in human T cells or hematopoietic progenitor cells [
209,
210]. It is anticipated that the editing efficiencies of base editors or PEs in human PSCs can also be improved with these delivery techniques.
In addition to modifications on editors or gRNAs, small molecules were also found to be able to manipulate editing efficiencies. Small molecules that can enhance the HDR activity, such as L755507 and Brefeldin A, also increase CRISPR-mediated HDR efficiencies in mouse ESCs and human non-pluripotent cells [
211]. Inhibitors targeting key components of NHEJ also increase the HDR rate in human non-pluripotent cells and mouse embryos [
212,
213,
214]. In addition, inhibition of ATM or ATR could enhance both knockout and knock-in efficiencies of Cas12a (Cpf1) in human PSCs, although whether it is applicable for Cas9 remains to be determined [
215]. Finally, histone deacetylase (HDAC) inhibitors can enhance Cas9-, CBE-, and ABE-mediated editing by increasing both the expression level of proteins and target accessibility in human non-pluripotent cells [
215]. Investigating these boosters and their influences on off-target effects as well as genome integrity in the context of human PSCs will benefit future applications.
6.2. Conditional Gene Editing
The major concern of performing gene editing in PSCs is that off-target edits will be carried to their differentiated progenies. One potential solution of that is to construct inactive editing components in PSCs which will be activated upon differentiation to perform editing in somatic (stem) cells that have limited longevity. This design can be achieved by putting editors and/or gRNAs under the control of specific promoters. However, most RNA polymerase III promoters used to drive gRNA expression are constitutively active. We recently established a novel PE, p2PE3, using RNA polymerase II promoters to drive the expression of pegRNA and sgRNA [
187] (
Figure 2c). The p2PE3 displays 2.1-fold higher editing efficiency compared to PE3, and can be combined with SV40LT and/or MLH1dn to further increase its editing efficiency in human PSCs [
187]. Using this system, the PE and pegRNA can be integrated as a cassette and put under the control of drug-inducible or lineage-specific promoters. This conditional editing strategy can also be employed by Cas9 or base editors to avoid undesired mutation in PSCs.
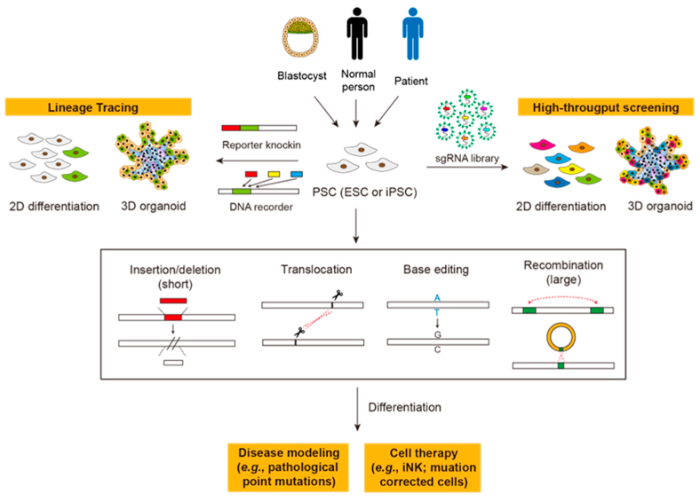
6.3. CRISPR-Cas as DNA Recorders of Cell Fates
PSCs are reliable in vitro models for differentiation, which involves progressive transitions of cellular states. General lineage tracing approaches only allow marking one or two states (e.g., the Cre or the Cre/Dre dual system). Recently, the CRISPR-Cas system has been employed to record signaling, cellular, or transcriptional events on DNA of eukaryotic cells (i.e., using DNA as the memory device) [
216,
217,
218,
219]. Among them, the “DNA typewriter” technique employs an elegant design, using sequential prime editing to capture different events in the happening order [
219]. With this technique, different events, such as transcription activation or signal transduction, can be encoded by different pegRNAs driven by specific promoters (i.e., the p2PE3 system mentioned before) built in PSCs. Upon 2D or 3D differentiation, the order of happened events can be recorded on the “DNA tape” in each cell and decoded by single-cell sequencing. This system could be a unique tool to reveal complex event histories during cell fate specification.
6.4. New Systems to Be Explored
In nature, there are still broad varieties of RNA-guided nucleases, transposases, and recombinases that remain unexplored and can be potentially employed as gene editing tools. For example, the Doudna group identified the CIRSPR-CasΦ system from the Biggiephage [
220], which is only half the size of Cas9 and has been employed for gene editing in plants [
221]. A more thorough investigation identified ~6000 phage CRISPR-Cas systems covering all six known CRISPR-Cas types [
222]. One of them, Casλ, was characterized and able to perform gene editing in HEK293T cells [
222]. By tracing the ancestor of Cas proteins, the Zhang and Siksnys groups identified three IS200/IS605 transposon-encoded proteins, IscB, IsrB, and TnpB, which are also RNA-guided DNA nucleases [
223,
224]. Both IscB and TnpB exhibit gene editing activity and can be incorporated in base editors with high efficiencies in human cells [
223,
224,
225,
226]. Surprisingly, two very recent studies suggest that TnpB homologs are widespread in eukaryotes [
227,
228]. Saito et al. and Jiang et al. characterized the RNA-guided DNA nuclease activity of the eukaryotic transposon-encoded Fanzor proteins. Both studies demonstrated that Fanzor proteins from different species can be reprogrammed for human genome engineering in HEK293T cells [
227,
228]. Those eukaryotic RNA-guided endonucleases not only have hypercompact sizes but also exhibit low cleavage activity on collateral nucleic acids. Since they are originated from eukaryotic cells, Fanzor proteins are expected to have great application potential in the future.
In addition to novel nucleases, the Zhang group also elucidated the transposition mechanism of a non–long terminal repeat (non-LTR) retrotransposon, the R2 retrotransposon. Non-LTR transposons are inserted into genomes by a mechanism called target-primed reverse transcription (TPRT), during which the target DNA sequence is nicked, priming the reverse transcription of retrotransposon RNA. The Zhang group resolved the structure of the silk moth R2Bm (LINE type) TPRT complex and elucidated the mechanism of how R2Bm recognizes its native target to initiate TPRT [
229]. Importantly, the Zhang group found that Cas9 can retarget R2 in vitro and initiate TPRT [
229]. Although the integration events have not yet been observed in vitro or in vivo, this finding suggests its future use as a site-specific insertion tool.
6.5. Conclusions
With the optimization of current tools and the discovery of new tools, it is predictable that “safe” gene editing in PSCs will be easier to perform in the future. Notably, since the specificity of gene editing in PSCs has to meet the highest criteria, those gene editors validated in PSCs can also be potentially applied to other gene- or cell-therapies. Undeniably, current PSC-based therapies still face concerns, such as the removal of residual undifferentiated cells as well as the immunogenicity and under-performance of PSC-derived cells. Gene editing could also be employed to resolve those issues by engineering cells for lower immunogenicity (e.g., removal of HLA) or high efficacies/functions (e.g., expression of stimulatory or sustaining cytokines). Considering PSCs’ unique merits of stability and unlimited quantity, it is worthy to further develop and validate gene editing tools for PSCs to facilitate their applications in both basic and translational research.