Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Helena Motaln | -- | 2548 | 2023-08-24 11:37:59 | | | |
| 2 | Dean Liu | -2 word(s) | 2546 | 2023-08-25 02:41:20 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Motaln, H.; Rogelj, B. c-Abl Tyrosine Kinase in Brain and Its Pathologies. Encyclopedia. Available online: https://encyclopedia.pub/entry/48428 (accessed on 19 June 2026).
Motaln H, Rogelj B. c-Abl Tyrosine Kinase in Brain and Its Pathologies. Encyclopedia. Available at: https://encyclopedia.pub/entry/48428. Accessed June 19, 2026.
Motaln, Helena, Boris Rogelj. "c-Abl Tyrosine Kinase in Brain and Its Pathologies" Encyclopedia, https://encyclopedia.pub/entry/48428 (accessed June 19, 2026).
Motaln, H., & Rogelj, B. (2023, August 24). c-Abl Tyrosine Kinase in Brain and Its Pathologies. In Encyclopedia. https://encyclopedia.pub/entry/48428
Motaln, Helena and Boris Rogelj. "c-Abl Tyrosine Kinase in Brain and Its Pathologies." Encyclopedia. Web. 24 August, 2023.
Copy Citation
Among kinases, non-receptor tyrosine kinase (Abelson kinase) c-Abl appears to be involved in both the normal development of neural tissue and the development of neurodegenerative pathologies when abnormally expressed or activated. However, exactly how c-Abl mediates the progression of neurodegeneration remains largely unexplored.
c-Abl (Abelson) tyrosine kinase
neurodegeneration
Alzheimer’s disease
1. Introduction
Despite all the progress in understanding neurodegeneration, researchers are still unable to close the knowledge gap about what exactly happens in brain cells just before the onset of neurodegenerative diseases like Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) or frontotemporal dementia (FTD). This is probably one of the reasons why there are still no effective treatments for all these diseases and why many targets identified so far have shown very limited neuroprotective effects in human studies [1]. Because current treatments are mostly symptomatic, researchers are urgently seeking novel neuroprotective agents and disease-modifying strategies that would slow or hopefully halt the progression of neurodegeneration altogether.
What several neurodegenerative diseases (ND) have in common is the death of various types of neurons, usually due to the formation of extracellular and/or intracellular protein inclusions that impair cellular processes, disbalance homeostasis and induce programmed cell death or apoptosis [1][2]. In many ND including ALS, the pathogenesis and death of motor neurons are also thought to be triggered by non-cell-autonomous mechanisms, since the conditioned medium from SOD1-mutant primary mouse astrocytes has been shown to cause death of exposed cultured motor neurons in vitro [3]. Moreover, the strong correlation between cognitive decline and synapse loss in several ND supports the idea that synaptic damage may indeed be one of the main pathogenic mechanisms underlying the development and progression of neurodegeneration [4]. However, considering that in AD, PD and FTD, in addition to the observed synaptic loss, defects in neurotransmitter activity, signaling efficiency, damage/repair systems, cell cycle, glial function, and neuroinflammatory processes have also been identified, a true major target of neurodegeneration may be the intracellular signaling machinery provided by the kinome [5]. Kinases are known to play critical roles in various cell signaling pathways [6] and have been confirmed to be dysregulated in a number of diseases, including neurodegeneration [7]. They provide a link between cell surface recognition events triggered by the binding of cell adhesion molecules, extracellular matrix, or other soluble factors (e.g., growth factors) and intracellular signaling pathways in neuronal cells [8]. Based on the observations of an inverse relationship between cancer and ND, a focus has been placed in the past on some of the cancer kinases to target intracellular signaling pathways at the intersection between the control of cellular metabolism and proliferation, inhibition of which was thought to halt neurodegeneration [5], but so far this inhibition has not shown an efficient therapeutic effect [9][10]. Nonetheless, therapeutics targeting kinases currently still account for approximately 50% of anticancer drug discovery efforts [7].
Non-receptor protein tyrosine kinases of the Src family (c-Src, c-Fyn, c-Yes, and c-Abl) are associated with ND as they are involved in axonal and dendritic outgrowth during central and peripheral nervous system development and regeneration [8]. In this context, aberrant c-Abl activation was shown to cause early neuroinflammation and loss of neurons in the forebrain of Niemann–Pick type C (NPC) transgenic mice [11], and increased c-Abl activation has been reported in neurodegenerative pathologies of PD, AD, ALS and FTD by researchers and others [11][12][13]. In the brains of patients with AD, c-Abl activity is associated with the formation of neuritic plaques and insoluble neurofibrillary tangles [14], whereas in FTD-FUS cases, increased c-Abl activity was associated with C-terminal Tyr phosphorylation of FUS protein and its aggregation in cortical neurons [12]. This suggests that abnormal activation of c-Abl may contribute to nonspecific posttranslational modifications of ND-related proteins, which may then promote the occurrence of features associated with ND, such as the accumulation of insoluble protein aggregates and impaired mitochondrial function, both of which are accompanied by synaptic damage.
Oxidative stress, the most likely trigger of abnormal kinome activation, has long been implicated in the pathogenesis of ND [15] and has been reported as a major cause of sporadic PD [16], where it is responsible for much of the dopaminergic neuronal damage [17]. The ubiquitously expressed non-receptor tyrosine kinase c-Abl is activated by oxidative stress and plays a role in oxidative stress-induced neuronal cell death [18][19]. There it is even considered an indicator of oxidative stress [16][17]. Selenocysteine insertion sequence-associating factors, adenosine, Arg kinase and c-Abl kinase are all potent Se-independent regulators of expression and activity of glutathione peroxidase-1 (GPX1) gene/protein, which plays a protective role in neuronal cells in coping with oxidative damage [20]. Activation of c-Abl, with few exceptions, mostly negatively affects enzymes involved in antioxidant defense. Still, c-Abl can be modified by S-glutathionylation, and this reversible modification leads to the down-regulation of its kinase activity [11][21]. Inversely, depending on the oxidation level in the cell, glutathione peroxidase can also be activated via phosphorylation at Tyr96 by c-Abl [22]. Although the constitutively active form of c-Abl, Bcr-Abl, has a long history in myeloid and lymphoblastic leukemia, aberrant activation of c-Abl has emerged as a link between various triggers of oxidative stress relevant to PD, AD, FTLD and α-synucleinopathies [12][15][23]. Inhibition of c-Abl kinase activity by small molecule compounds used in the clinic to treat human leukemia showed neuroprotective effects in cell and animal models of PD [24]. Unfortunately, to date, several c-Abl kinase inhibitors have shown only sub-threshold efficacy in clinical trials [9][10], most likely due to limited knowledge of c-Abl signaling. Therefore, researchers review the functions and effects of c-Abl in neuronal cells discovered to date to demonstrate how different aspects of c-Abl signaling contribute to the progression of neurodegenerative diseases. See Figure 1 and the following sections for an explanation.
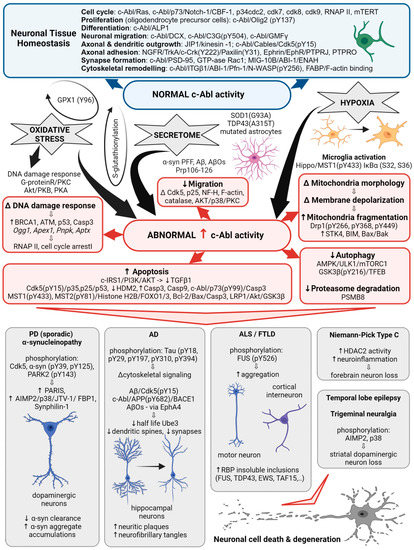
Figure 1. c-Abl signaling involved in multiple cellular processes. Schematic illustrating the different Abl signaling pathways discussed in the following text and highlighting the correlation with the abnormal processes associated with neurodegenerative diseases.
2. The Structure of c-Abl and Its Role in Neurodegenerative Diseases
Initially, the non-receptor tyrosine kinase c-Abl was identified as a protooncogene activated in a subset of human leukemias [25], yet quite soon it was associated with neurodegeneration. The c-Abl kinase has a complex structure consisting of multiple domains and motifs that are also found in other signal-transducing proteins [25] and have been reviewed in detail elsewhere [26]. Protein-protein interaction screens of a phage expression library have identified proteins that interact with specific domains of c-Abl and can be termed regulators or effectors of c-Abl activity. In this way, SH3-domain-containing proteins, amphiphysin-like protein 1 (ALP1) and amphiphysin are proposed to interact with the c-Abl carboxyl terminus to regulate its role in cell differentiation in vitro and in vivo [25]. Expression of ALP1 leads to the morphological transformation of NIH 3T3 fibroblasts in a c-Abl-dependent manner that involves remodeling of the cytoskeleton [25]. Moreover, increased c-Abl activity was detected in oligodendrocyte progenitor cells, which are essential for myelination during central nervous system development. In these, c-Abl-mediated phosphorylation of the transcription factor Olig2 was confirmed indispensable for the proliferation of oligodendrocyte progenitor cells [27]. Yet the homology of the yeast proteins Rvs167 and Rvs161 with the amino terminus of c-Abl and abnormal activation of c-Abl also suggest that it is involved in cell cycle arrest [25], neuroinflammation [28], and may cause neuronal death via activation of apoptotic signaling pathways [29]. Table 1 summarizes the status of c-Abl expression and activity detected in ND.
In PD, abnormally increased c-Abl activity is associated with the accumulation of pathogenic α-synuclein (α-syn) [30]. Increased expression and activation of c-Abl has been found in mouse models of PD and AD and in neuronal cultures in response to inclusions formation and oxidative stress. Overexpression of active c-Abl in mouse neurons leads to neurodegeneration and neuroinflammation [31]. Levels and activity of c-Abl are greatly increased in the brain tissue of patients with PD [32][33][34]. In dopaminergic neurons, this is accompanied by increased phosphorylation of c-Abl protein substrates, such as α-syn and the E3 ubiquitin ligase, parkin [24][35]. In animal models, the use of different c-Abl inhibitors has been shown to improve motor behavior in animals and prevent loss of dopaminergic neurons [1][35], while the inhibitors Nilotinib and Radontinib even showed improvement in motor and cognitive symptoms in PD patients [35]. The expression of c-Abl is increased in trigeminal neuralgia, where it is accompanied by the loss of dopamine neurons in the striatum via aminoacyl-tRNA synthetase-interacting multifunctional protein type2 (AIMP2, p38) activation [28]. In PD models, c-Abl inhibitors reduce phosphorylation of Cdk5, decrease phosphorylation and clearance of α-syn and parkin, and decrease levels of several parkin substrates such as zinc finger protein 746 (PARIS), AIMP2, fuse-binding protein 1 (FBP1), and synphilin-1 [17][32][36]. Radotinib has even been demonstrated to protect primary cortical neurons from toxic cell death induced by c-Abl activation with preformed α-syn fibrils and to reduce Lewy bodies/Lewy neurites-like pathology [35]. Overall, increased activation of c-Abl through parkin inactivation, accumulation of its toxic substrate AIMP2, α-syn aggregation, and impaired autophagy are shown to be associated with neurodegenerative processes of PD.
In AD, c-Abl plays a role in the development of Tau pathology by regulating cytoskeletal signaling cascades. Immunocytochemical studies show that c-Abl is associated with both neuritic plaques and neurofibrillary tangles in the brains of patients with AD. c-Abl interacts directly with Tau and phosphorylates it at tyrosine 394 [23][36], which has a regulatory effect on normal Tau-related processes, including microtubule assembly and axonal transport, and to trigger aggregation of Tau into paired helical filaments [26]. Neuronal spine pathology is associated with the early onset of AD. Amyloid beta oligomers (AβOs) are known to induce synaptotoxicity, leading to synaptic dysfunction/loss and the reduction in dendritic spine density that underlies cognitive defects [4]. c-Abl was activated in neurons exposed to AβOs and in the brains of patients with AD. Inhibition of active c-Abl ameliorated all AβOs-induced synaptic changes [37] and cognitive deficits in the AD mouse model [4]. AβOs induction of c-Abl signaling appears to involve the tyrosine kinase ephrin receptor A4 (EphA4) [4] and decreases the number of mushroom spines in c-Abl knockout neurons, while preserving the populations of immature stubby, filopodia spines, suggesting that c-Abl deficiency increases the population of immature spines and decreases AβOs-induced synapse elimination [37].
Table 1. c-Abl expression and activity in disease.
| Status | Detected in Patients and Cell or Animal Models | Reference |
|---|---|---|
| Increased c-Abl expression |
|
[11][15][23][24] [38][39] [9][40] [9] [41] [23][31] [42] [43] [44] [45] |
| Increased c-Abl activity |
|
[32][34][46][47] [48] [12] [47] [49] [50] [28] [30] [11] [41] [51] [52] [33] |
| c-Abl deficiency |
|
[37] [40] [46] |
In ALS, increased c-Abl expression was found in motoneurons [13]. A phenotypic screen of motor neurons derived from induced pluripotent stem cells (iPSCs) from ALS patients with SOD1 mutation revealed that more than half of the tested drugs that stop neuronal cell death target the Src/c-Abl pathway. The Src/c-Abl inhibitors increased the survival of iPSC-derived motor neurons from ALS patients in vitro, and siRNA knockdowns of c-Src or c-Abl prevented their degeneration. Likewise, Bosutinib inhibitor increased in vitro survival of iPSC-derived motor neurons from patients with sporadic or familial forms of ALS, caused by mutations in the TAR DNA binding protein (TDP-43) or repeat expansions in the C9orf72 gene [53]. Moreover, conditioned media from primary mouse astrocytes expressing either mutant human SOD1(G93A) or TDP43(A315T), but not from SOD1(WT) astrocytes, increased c-Abl activity in rat motoneurons, interneurons and glial cells in vitro, which was detected 60 min after exposure, and resulted in neuron death within days. This effect of the conditioned media was prevented by the use of the c-Abl inhibitor Imatinib, blockers of Na channels (spermidine, mexiletine, or riluzole), and antioxidants (Trolox, esculetin, or tiron) [3].
Finally, increased c-Abl activity has been found in other ND as well. This way increased c-Abl activity was noted in cortical neurons of FTLD-FUS patients [12], whereas both total and phosphorylated c-Abl were found upregulated in the temporal neocortex of patients with temporal lobe epilepsy compared to nonepileptic controls [41]. In the temporal neocortex of model rats treated with pilocarpine, upregulation of total and phosphorylated c-Abl begins 6 h after seizures, with relatively high levels persisting for 60 days, whereas in the hippocampus, elevated c-Abl levels persist for 30 days after seizures and then return to normal [41]. Given the evidence of c-Abl activation in the brain of patients with various ND, improved targeting of its signaling may indeed prove advantageous for novel treatment designs.
3. The Role of c-Abl in Brain Injuries
In addition to ND, c-Abl also appears to promote neuronal cell death in brain injury caused by hypoxia and cerebral ischemia [51]. In particular, cerebral ischemia-reperfusion injury represents a major public health problem that causes high rates of disability and death in adults [54]. Under hypoxic conditions, the normal development and migration of brain cells can be severely impaired, and c-Abl appears to be involved in processes leading to cell death. For example, increased c-Abl protein levels were observed in rat pups exposed to intermittent hypoxia during embryonic development, resulting in a delay in neuronal migration early in the postpartum period. The downstream targets of c-Abl: Cdk5, p25, and the cytoskeletal elements neurofilament H, F-actin, and catalase were all found altered [55]. Likewise, computational analyses of phosphoprotein datasets covering the response of sensory neurons to axonal injury identified 400 redundant axonal signaling networks, among which the signaling hub proteins c-Abl, AKT, p38, and protein kinase C, were overrepresented [56]. Moreover, endogenous c-Abl protein levels and neuronal apoptosis also increase 24 h after subarachnoid hemorrhage. This could be inhibited with c-Abl inhibitors decreasing cleavage of caspase-3 and enhancing the phosphorylation of Akt and glycogen synthase kinase (GSK)3β [51].
Activation of microglia also plays a role in the alteration of the neuronal microenvironment caused by ischemic stroke. c-Abl has been found to be involved in the mechanism underlying microglial activation and subsequent oxidative stress-induced cell death of primary neurons. c-Abl was found to phosphorylate Hippo/MST1 protein kinase at Y433, which increases its activity for phosphorylation of IκBα at residues S32 and S36, and lead to microglial activation [54]. Oxidative stress in brain cells can also be unknowingly induced by the use of iron-oxide nanoparticles as therapeutics or for supplemental intake in iron deficiency. Apparently, 24-h exposure of human SH-SY5Y neuroblastoma cells to 10 μg/mL of 10- and 30-nm iron oxide nanoparticles not only decreases cellular dopamine content by 50% but also increases and activates c-Abl and neuronal α-Syn expression. In mice exposed to these nanoparticles, the number of active mitochondria in neuronal cells and striatal dopamine and its metabolites decrease, and neuropathological damage to neuronal cell bodies, dopaminergic terminal, and neuronal vasculature occurs [57].
References
- Abushouk, A.I.; Negida, A.; Elshenawy, R.A.; Zein, H.; Hammad, A.M.; Menshawy, A.; Mohamed, W.M.Y. C-Abl Inhibition; A Novel Therapeutic Target for Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2018, 17, 14–21.
- La Barbera, L.; Vedele, F.; Nobili, A.; Krashia, P.; Spoleti, E.; Latagliata, E.C.; Cutuli, D.; Cauzzi, E.; Marino, R.; Viscomi, M.T.; et al. Nilotinib Restores Memory Function by Preventing Dopaminergic Neuron Degeneration in a Mouse Model of Alzheimer’s Disease. Prog. Neurobiol. 2021, 202, 102031.
- Rojas, F.; Gonzalez, D.; Cortes, N.; Ampuero, E.; Hernández, D.E.; Fritz, E.; Abarzua, S.; Martinez, A.; Elorza, A.A.; Alvarez, A.; et al. Reactive Oxygen Species Trigger Motoneuron Death in Non-Cell-Autonomous Models of ALS through Activation of c-Abl Signaling. Front. Cell. Neurosci. 2015, 9, 203.
- Vargas, L.M.; Cerpa, W.; Muñoz, F.J.; Zanlungo, S.; Alvarez, A.R. Amyloid-β Oligomers Synaptotoxicity: The Emerging Role of EphA4/c-Abl Signaling in Alzheimer’s Disease. Biochim. Biophys. Acta Mol. basis Dis. 2018, 1864, 1148–1159.
- Fagiani, F.; Lanni, C.; Racchi, M.; Govoni, S. Targeting Dementias through Cancer Kinases Inhibition. Alzheimer’s Dement. 2020, 6, e12044.
- Ranganathan, S.; Dasmeh, P.; Furniss, S.; Shakhnovich, E. Phosphorylation Sites Are Evolutionary Checkpoints against Liquid-Solid Transition in Protein Condensates. Proc. Natl. Acad. Sci. USA 2023, 120, e2215828120.
- Albanese, S.K.; Parton, D.L.; Işık, M.; Rodríguez-Laureano, L.; Hanson, S.M.; Behr, J.M.; Gradia, S.; Jeans, C.; Levinson, N.M.; Seeliger, M.A.; et al. An Open Library of Human Kinase Domain Constructs for Automated Bacterial Expression. Biochemistry 2018, 57, 4675–4689.
- Maness, P.F. Nonreceptor Protein Tyrosine Kinases Associated with Neuronal Development. Dev. Neurosci. 1992, 14, 257–270.
- Lopez-Cuina, M.; Guerin, P.A.; Canron, M.-H.; Delamarre, A.; Dehay, B.; Bezard, E.; Meissner, W.G.; Fernagut, P.-O. Nilotinib Fails to Prevent Synucleinopathy and Cell Loss in a Mouse Model of Multiple System Atrophy. Mov. Disord. 2020, 35, 1163–1172.
- Hung, A.Y.; Schwarzschild, M.A. Approaches to Disease Modification for Parkinson’s Disease: Clinical Trials and Lessons Learned. Neurother. J. Am. Soc. Exp. Neurother. 2020, 17, 1393–1405.
- Gonfloni, S.; Maiani, E.; Di Bartolomeo, C.; Diederich, M.; Cesareni, G. Oxidative Stress, DNA Damage, and c-Abl Signaling: At the Crossroad in Neurodegenerative Diseases? Int. J. Cell Biol. 2012, 2012, 683097.
- Motaln, H.; Čerček, U.; Yamoah, A.; Tripathi, P.; Aronica, E.; Goswami, A.; Rogelj, B. Abl Kinase-Mediated FUS Tyr526 Phosphorylation Alters Nucleocytoplasmic FUS Localization in FTLD-FUS. Brain 2023, awad130.
- Katsumata, R.; Ishigaki, S.; Katsuno, M.; Kawai, K.; Sone, J.; Huang, Z.; Adachi, H.; Tanaka, F.; Urano, F.; Sobue, G. C-Abl Inhibition Delays Motor Neuron Degeneration in the G93A Mouse, an Animal Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e46185.
- Feng, L.; Sun, Z.-G.; Liu, Q.-W.; Ma, T.; Xu, Z.-P.; Feng, Z.-G.; Yuan, W.-X.; Zhang, H.; Xu, L.-H. Propofol Inhibits the Expression of Abelson Nonreceptor Tyrosine Kinase without Affecting Learning or Memory Function in Neonatal Rats. Brain Behav. 2020, 10, e01810.
- Brahmachari, S.; Karuppagounder, S.S.; Ge, P.; Lee, S.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. C-Abl and Parkinson’s Disease: Mechanisms and Therapeutic Potential. J. Parkinsons. Dis. 2017, 7, 589–601.
- Wu, R.; Chen, H.; Ma, J.; He, Q.; Huang, Q.; Liu, Q.; Li, M.; Yuan, Z. C-Abl-P38α Signaling Plays an Important Role in MPTP-Induced Neuronal Death. Cell Death Differ. 2016, 23, 542–552.
- Imam, S.Z.; Trickler, W.; Kimura, S.; Binienda, Z.K.; Paule, M.G.; Slikker, W.J.; Li, S.; Clark, R.A.; Ali, S.F. Neuroprotective Efficacy of a New Brain-Penetrating C-Abl Inhibitor in a Murine Parkinson’s Disease Model. PLoS ONE 2013, 8, e65129.
- Karuppagounder, S.S.; Wang, H.; Kelly, T.; Rush, R.; Nguyen, R.; Bisen, S.; Yamashita, Y.; Sloan, N.; Dang, B.; Sigmon, A.; et al. The C-Abl Inhibitor IkT-148009 Suppresses Neurodegeneration in Mouse Models of Heritable and Sporadic Parkinson’s Disease. Sci. Transl. Med. 2023, 15, eabp9352.
- Marín, T.; Valls, C.; Jerez, C.; Huerta, T.; Elgueta, D.; Vidal, R.L.; Alvarez, A.R.; Cancino, G.I. The C-Abl/P73 Pathway Induces Neurodegeneration in a Parkinson’s Disease Model. IBRO Neurosci. Rep. 2022, 13, 378–387.
- Lei, X.G.; Cheng, W.-H.; McClung, J.P. Metabolic Regulation and Function of Glutathione Peroxidase-1. Annu. Rev. Nutr. 2007, 27, 41–61.
- Leonberg, A.K.; Chai, Y.-C. The Functional Role of Cysteine Residues for C-Abl Kinase Activity. Mol. Cell. Biochem. 2007, 304, 207–212.
- Cao, C.; Leng, Y.; Huang, W.; Liu, X.; Kufe, D. Glutathione Peroxidase 1 Is Regulated by the C-Abl and Arg Tyrosine Kinases. J. Biol. Chem. 2003, 278, 39609–39614.
- Schlatterer, S.D.; Tremblay, M.A.; Acker, C.M.; Davies, P. Neuronal C-Abl Overexpression Leads to Neuronal Loss and Neuroinflammation in the Mouse Forebrain. J. Alzheimers. Dis. 2011, 25, 119–133.
- Lindholm, D.; Pham, D.D.; Cascone, A.; Eriksson, O.; Wennerberg, K.; Saarma, M. C-Abl Inhibitors Enable Insights into the Pathophysiology and Neuroprotection in Parkinson’s Disease. Front. Aging Neurosci. 2016, 8, 254.
- Kadlec, L.; Pendergast, A.M. The Amphiphysin-like Protein 1 (ALP1) Interacts Functionally with the CABL Tyrosine Kinase and May Play a Role in Cytoskeletal Regulation. Proc. Natl. Acad. Sci. USA 1997, 94, 12390–12395.
- Estrada, L.D.; Zanlungo, S.M.; Alvarez, A.R. C-Abl Tyrosine Kinase Signaling: A New Player in AD Tau Pathology. Curr. Alzheimer Res. 2011, 8, 643–651.
- Zhang, J.; Sun, J.-G.; Xing, X.; Wu, R.; Zhou, L.; Zhang, Y.; Yuan, F.; Wang, S.; Yuan, Z. C-Abl-Induced Olig2 Phosphorylation Regulates the Proliferation of Oligodendrocyte Precursor Cells. Glia 2022, 70, 1084–1099.
- Fu, J.; Mu, G.; Qiu, L.; Zhao, J.; Ou, C. C-Abl-P38α Signaling Pathway Mediates Dopamine Neuron Loss in Trigeminal Neuralgia. Mol. Pain 2020, 16, 1744806920930855.
- Dash, D.; Goyal, V. Anticancer Drugs for Parkinson’s Disease: Is It a Ray of Hope or Only Hype? Ann. Indian Acad. Neurol. 2019, 22, 13–16.
- Karim, M.R.; Liao, E.E.; Kim, J.; Meints, J.; Martinez, H.M.; Pletnikova, O.; Troncoso, J.C.; Lee, M.K. α-Synucleinopathy Associated c-Abl Activation Causes P53-Dependent Autophagy Impairment. Mol. Neurodegener. 2020, 15, 27.
- Schlatterer, S.D.; Acker, C.M.; Davies, P. C-Abl in Neurodegenerative Disease. J. Mol. Neurosci. 2011, 45, 445–452.
- Zhou, Z.-H.; Wu, Y.-F.; Wang, X.-M.; Han, Y.-Z. The C-Abl Inhibitor in Parkinson Disease. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2017, 38, 547–552.
- Ren, Y.; Chen, J.; Wu, X.; Gui, C.; Mao, K.; Zou, F.; Li, W. Role of C-Abl-GSK3β Signaling in MPP+-Induced Autophagy-Lysosomal Dysfunction. Toxicol. Sci. 2018, 165, 232–243.
- Karuppagounder, S.S.; Brahmachari, S.; Lee, Y.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. The C-Abl Inhibitor, Nilotinib, Protects Dopaminergic Neurons in a Preclinical Animal Model of Parkinson’s Disease. Sci. Rep. 2014, 4, 4874.
- Lee, S.; Kim, S.; Park, Y.J.; Yun, S.P.; Kwon, S.-H.; Kim, D.; Kim, D.Y.; Shin, J.S.; Cho, D.J.; Lee, G.Y.; et al. The C-Abl Inhibitor, Radotinib HCl, Is Neuroprotective in a Preclinical Parkinson’s Disease Mouse Model. Hum. Mol. Genet. 2018, 27, 2344–2356.
- Hebron, M.L.; Lonskaya, I.; Olopade, P.; Selby, S.T.; Pagan, F.; Moussa, C.E.-H. Tyrosine Kinase Inhibition Regulates Early Systemic Immune Changes and Modulates the Neuroimmune Response in α-Synucleinopathy. J. Clin. Cell. Immunol. 2014, 5, 259.
- Gutierrez, D.A.; Vargas, L.M.; Chandia-Cristi, A.; de la Fuente, C.; Leal, N.; Alvarez, A.R. C-Abl Deficiency Provides Synaptic Resiliency Against Aβ-Oligomers. Front. Cell. Neurosci. 2019, 13, 526.
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E.-H. Nilotinib Reverses Loss of Dopamine Neurons and Improves Motor Behavior via Autophagic Degradation of α-Synuclein in Parkinson’s Disease Models. Hum. Mol. Genet. 2013, 22, 3315–3328.
- Mahul-Mellier, A.-L.; Fauvet, B.; Gysbers, A.; Dikiy, I.; Oueslati, A.; Georgeon, S.; Lamontanara, A.J.; Bisquertt, A.; Eliezer, D.; Masliah, E.; et al. C-Abl Phosphorylates α-Synuclein and Regulates Its Degradation: Implication for α-Synuclein Clearance and Contribution to the Pathogenesis of Parkinson’s Disease. Hum. Mol. Genet. 2014, 23, 2858–2879.
- Brahmachari, S.; Ge, P.; Lee, S.H.; Kim, D.; Karuppagounder, S.S.; Kumar, M.; Mao, X.; Shin, J.H.; Lee, Y.; Pletnikova, O.; et al. Activation of Tyrosine Kinase C-Abl Contributes to α-Synuclein-Induced Neurodegeneration. J. Clin. Investig. 2016, 126, 2970–2988.
- Chen, L.; Wang, Z.; Tang, B.; Fang, M.; Li, J.; Chen, G.; Wang, X. Altered Expression of C-Abl in Patients with Epilepsy and in a Rat Model. Synapse 2014, 68, 306–316.
- Allen, K.M.; Walsh, C.A. Genes That Regulate Neuronal Migration in the Cerebral Cortex. Epilepsy Res. 1999, 36, 143–154.
- Yáñez, M.J.; Belbin, O.; Estrada, L.D.; Leal, N.; Contreras, P.S.; Lleó, A.; Burgos, P.V.; Zanlungo, S.; Alvarez, A.R. C-Abl Links APP-BACE1 Interaction Promoting APP Amyloidogenic Processing in Niemann-Pick Type C Disease. Biochim. Biophys. Acta 2016, 1862, 2158–2167.
- Montecino, F.; González, N.; Blanco, N.; Ramírez, M.J.; González-Martín, A.; Alvarez, A.R.; Olguín, H. C-Abl Kinase Is Required for Satellite Cell Function Through Pax7 Regulation. Front. Cell Dev. Biol. 2021, 9, 606403.
- Perez de Arce, K.; Varela-Nallar, L.; Farias, O.; Cifuentes, A.; Bull, P.; Couch, B.A.; Koleske, A.J.; Inestrosa, N.C.; Alvarez, A.R. Synaptic Clustering of PSD-95 Is Regulated by c-Abl through Tyrosine Phosphorylation. J. Neurosci. 2010, 30, 3728–3738.
- Ko, H.S.; Lee, Y.; Shin, J.-H.; Karuppagounder, S.S.; Gadad, B.S.; Koleske, A.J.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M. Phosphorylation by the C-Abl Protein Tyrosine Kinase Inhibits Parkin’s Ubiquitination and Protective Function. Proc. Natl. Acad. Sci. USA 2010, 107, 16691–16696.
- Imam, S.Z.; Zhou, Q.; Yamamoto, A.; Valente, A.J.; Ali, S.F.; Bains, M.; Roberts, J.L.; Kahle, P.J.; Clark, R.A.; Li, S. Novel Regulation of Parkin Function through C-Abl-Mediated Tyrosine Phosphorylation: Implications for Parkinson’s Disease. J. Neurosci. 2011, 31, 157–163.
- Lebouvier, T.; Scales, T.M.E.; Williamson, R.; Noble, W.; Duyckaerts, C.; Hanger, D.P.; Reynolds, C.H.; Anderton, B.H.; Derkinderen, P. The Microtubule-Associated Protein Tau Is Also Phosphorylated on Tyrosine. J. Alzheimers. Dis. 2009, 18, 1–9.
- Yañez, M.J.; Campos, F.; Marín, T.; Klein, A.D.; Futerman, A.H.; Alvarez, A.R.; Zanlungo, S. C-Abl Activates RIPK3 Signaling in Gaucher Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166089.
- Zhou, L.; Zhang, Q.; Zhang, P.; Sun, L.; Peng, C.; Yuan, Z.; Cheng, J. C-Abl-Mediated Drp1 Phosphorylation Promotes Oxidative Stress-Induced Mitochondrial Fragmentation and Neuronal Cell Death. Cell Death Dis. 2017, 8, e3117.
- Yan, C.; Yu, H.; Liu, Y.; Wu, P.; Wang, C.; Zhao, H.; Yang, K.; Shao, Q.; Zhong, Y.; Zhao, W.; et al. C-Abl Tyrosine Kinase-Mediated Neuronal Apoptosis in Subarachnoid Hemorrhage by Modulating the LRP-1-Dependent Akt/GSK3β Survival Pathway. J. Mol. Neurosci. 2021, 71, 2514–2525.
- Gonzalez-Zuñiga, M.; Contreras, P.S.; Estrada, L.D.; Chamorro, D.; Villagra, A.; Zanlungo, S.; Seto, E.; Alvarez, A.R. C-Abl Stabilizes HDAC2 Levels by Tyrosine Phosphorylation Repressing Neuronal Gene Expression in Alzheimer’s Disease. Mol. Cell 2014, 56, 163–173.
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The Src/c-Abl Pathway Is a Potential Therapeutic Target in Amyotrophic Lateral Sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962.
- Zhao, S.; Yin, J.; Zhou, L.; Yan, F.; He, Q.; Huang, L.; Peng, S.; Jia, J.; Cheng, J.; Chen, H.; et al. Hippo/MST1 Signaling Mediates Microglial Activation Following Acute Cerebral Ischemia-Reperfusion Injury. Brain. Behav. Immun. 2016, 55, 236–248.
- Zechel, J.L.; Gamboa, J.L.; Peterson, A.G.; Puchowicz, M.A.; Selman, W.R.; Lust, W.D. Neuronal Migration Is Transiently Delayed by Prenatal Exposure to Intermittent Hypoxia. Birth Defects Res. B Dev. Reprod. Toxicol. 2005, 74, 287–299.
- Michaelevski, I.; Segal-Ruder, Y.; Rozenbaum, M.; Medzihradszky, K.F.; Shalem, O.; Coppola, G.; Horn-Saban, S.; Ben-Yaakov, K.; Dagan, S.Y.; Rishal, I.; et al. Signaling to Transcription Networks in the Neuronal Retrograde Injury Response. Sci. Signal. 2010, 3, ra53.
- Imam, S.Z.; Lantz-McPeak, S.M.; Cuevas, E.; Rosas-Hernandez, H.; Liachenko, S.; Zhang, Y.; Sarkar, S.; Ramu, J.; Robinson, B.L.; Jones, Y.; et al. Iron Oxide Nanoparticles Induce Dopaminergic Damage: In Vitro Pathways and In Vivo Imaging Reveals Mechanism of Neuronal Damage. Mol. Neurobiol. 2015, 52, 913–926.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
612
Revisions:
2 times
(View History)
Update Date:
25 Aug 2023
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No