Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Helena Motaln and Version 2 by Dean Liu.
Among kinases, non-receptor tyrosine kinase (Abelson kinase) c-Abl appears to be involved in both the normal development of neural tissue and the development of neurodegenerative pathologies when abnormally expressed or activated. However, exactly how c-Abl mediates the progression of neurodegeneration remains largely unexplored.
- c-Abl (Abelson) tyrosine kinase
- neurodegeneration
- Alzheimer’s disease
1. Introduction
Despite all the progress in understanding neurodegeneration, reswearchers are still unable to close the knowledge gap about what exactly happens in brain cells just before the onset of neurodegenerative diseases like Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) or frontotemporal dementia (FTD). This is probably one of the reasons why there are still no effective treatments for all these diseases and why many targets identified so far have shown very limited neuroprotective effects in human studies [1]. Because current treatments are mostly symptomatic, researchers are urgently seeking novel neuroprotective agents and disease-modifying strategies that would slow or hopefully halt the progression of neurodegeneration altogether.
What several neurodegenerative diseases (ND) have in common is the death of various types of neurons, usually due to the formation of extracellular and/or intracellular protein inclusions that impair cellular processes, disbalance homeostasis and induce programmed cell death or apoptosis [1][2][1,2]. In many ND including ALS, the pathogenesis and death of motor neurons are also thought to be triggered by non-cell-autonomous mechanisms, since the conditioned medium from SOD1-mutant primary mouse astrocytes has been shown to cause death of exposed cultured motor neurons in vitro [3]. Moreover, the strong correlation between cognitive decline and synapse loss in several ND supports the idea that synaptic damage may indeed be one of the main pathogenic mechanisms underlying the development and progression of neurodegeneration [4]. However, considering that in AD, PD and FTD, in addition to the observed synaptic loss, defects in neurotransmitter activity, signaling efficiency, damage/repair systems, cell cycle, glial function, and neuroinflammatory processes have also been identified, a true major target of neurodegeneration may be the intracellular signaling machinery provided by the kinome [5]. Kinases are known to play critical roles in various cell signaling pathways [6] and have been confirmed to be dysregulated in a number of diseases, including neurodegeneration [7]. They provide a link between cell surface recognition events triggered by the binding of cell adhesion molecules, extracellular matrix, or other soluble factors (e.g., growth factors) and intracellular signaling pathways in neuronal cells [8]. Based on the observations of an inverse relationship between cancer and ND, a focus has been placed in the past on some of the cancer kinases to target intracellular signaling pathways at the intersection between the control of cellular metabolism and proliferation, inhibition of which was thought to halt neurodegeneration [5], but so far this inhibition has not shown an efficient therapeutic effect [9][10][9,10]. Nonetheless, therapeutics targeting kinases currently still account for approximately 50% of anticancer drug discovery efforts [7].
Non-receptor protein tyrosine kinases of the Src family (c-Src, c-Fyn, c-Yes, and c-Abl) are associated with ND as they are involved in axonal and dendritic outgrowth during central and peripheral nervous system development and regeneration [8]. In this context, aberrant c-Abl activation was shown to cause early neuroinflammation and loss of neurons in the forebrain of Niemann–Pick type C (NPC) transgenic mice [11], and increased c-Abl activation has been reported in neurodegenerative pathologies of PD, AD, ALS and FTD by reusearchers and others [11][12][13][11,12,13]. In the brains of patients with AD, c-Abl activity is associated with the formation of neuritic plaques and insoluble neurofibrillary tangles [14], whereas in FTD-FUS cases, increased c-Abl activity was associated with C-terminal Tyr phosphorylation of FUS protein and its aggregation in cortical neurons [12]. This suggests that abnormal activation of c-Abl may contribute to nonspecific posttranslational modifications of ND-related proteins, which may then promote the occurrence of features associated with ND, such as the accumulation of insoluble protein aggregates and impaired mitochondrial function, both of which are accompanied by synaptic damage.
Oxidative stress, the most likely trigger of abnormal kinome activation, has long been implicated in the pathogenesis of ND [15] and has been reported as a major cause of sporadic PD [16], where it is responsible for much of the dopaminergic neuronal damage [17]. The ubiquitously expressed non-receptor tyrosine kinase c-Abl is activated by oxidative stress and plays a role in oxidative stress-induced neuronal cell death [18][19][18,19]. There it is even considered an indicator of oxidative stress [16][17][16,17]. Selenocysteine insertion sequence-associating factors, adenosine, Arg kinase and c-Abl kinase are all potent Se-independent regulators of expression and activity of glutathione peroxidase-1 (GPX1) gene/protein, which plays a protective role in neuronal cells in coping with oxidative damage [20]. Activation of c-Abl, with few exceptions, mostly negatively affects enzymes involved in antioxidant defense. Still, c-Abl can be modified by S-glutathionylation, and this reversible modification leads to the down-regulation of its kinase activity [11][21][11,21]. Inversely, depending on the oxidation level in the cell, glutathione peroxidase can also be activated via phosphorylation at Tyr96 by c-Abl [22]. Although the constitutively active form of c-Abl, Bcr-Abl, has a long history in myeloid and lymphoblastic leukemia, aberrant activation of c-Abl has emerged as a link between various triggers of oxidative stress relevant to PD, AD, FTLD and α-synucleinopathies [12][15][23][12,15,23]. Inhibition of c-Abl kinase activity by small molecule compounds used in the clinic to treat human leukemia showed neuroprotective effects in cell and animal models of PD [24]. Unfortunately, to date, several c-Abl kinase inhibitors have shown only sub-threshold efficacy in clinical trials [9][10][9,10], most likely due to limited knowledge of c-Abl signaling. Therefore, heresearchers we review the functions and effects of c-Abl in neuronal cells discovered to date to demonstrate how different aspects of c-Abl signaling contribute to the progression of neurodegenerative diseases. See Figure 1 and the following sections for an explanation.
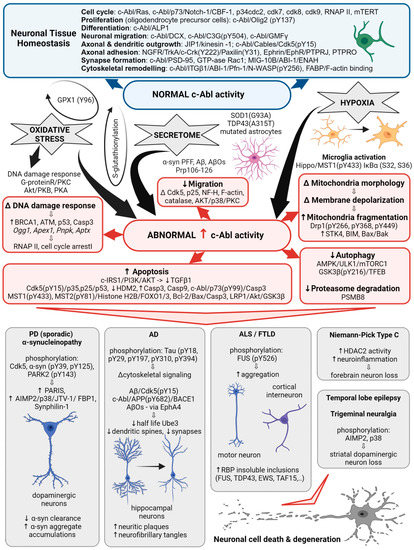
Figure 1. c-Abl signaling involved in multiple cellular processes. Schematic illustrating the different Abl signaling pathways discussed in the following text and highlighting the correlation with the abnormal processes associated with neurodegenerative diseases.
2. The Structure of c-Abl and Its Role in Neurodegenerative Diseases
Initially, the non-receptor tyrosine kinase c-Abl was identified as a protooncogene activated in a subset of human leukemias [25], yet quite soon it was associated with neurodegeneration. The c-Abl kinase has a complex structure consisting of multiple domains and motifs that are also found in other signal-transducing proteins [25] and have been reviewed in detail elsewhere [26]. Protein-protein interaction screens of a phage expression library have identified proteins that interact with specific domains of c-Abl and can be termed regulators or effectors of c-Abl activity. In this way, SH3-domain-containing proteins, amphiphysin-like protein 1 (ALP1) and amphiphysin are proposed to interact with the c-Abl carboxyl terminus to regulate its role in cell differentiation in vitro and in vivo [25]. Expression of ALP1 leads to the morphological transformation of NIH 3T3 fibroblasts in a c-Abl-dependent manner that involves remodeling of the cytoskeleton [25]. Moreover, increased c-Abl activity was detected in oligodendrocyte progenitor cells, which are essential for myelination during central nervous system development. In these, c-Abl-mediated phosphorylation of the transcription factor Olig2 was confirmed indispensable for the proliferation of oligodendrocyte progenitor cells [27]. Yet the homology of the yeast proteins Rvs167 and Rvs161 with the amino terminus of c-Abl and abnormal activation of c-Abl also suggest that it is involved in cell cycle arrest [25], neuroinflammation [28], and may cause neuronal death via activation of apoptotic signaling pathways [29]. Table 1 summarizes the status of c-Abl expression and activity detected in ND.
In PD, abnormally increased c-Abl activity is associated with the accumulation of pathogenic α-synuclein (α-syn) [30]. Increased expression and activation of c-Abl has been found in mouse models of PD and AD and in neuronal cultures in response to inclusions formation and oxidative stress. Overexpression of active c-Abl in mouse neurons leads to neurodegeneration and neuroinflammation [31]. Levels and activity of c-Abl are greatly increased in the brain tissue of patients with PD [32][33][34][32,33,34]. In dopaminergic neurons, this is accompanied by increased phosphorylation of c-Abl protein substrates, such as α-syn and the E3 ubiquitin ligase, parkin [24][35][24,35]. In animal models, the use of different c-Abl inhibitors has been shown to improve motor behavior in animals and prevent loss of dopaminergic neurons [1][35][1,35], while the inhibitors Nilotinib and Radontinib even showed improvement in motor and cognitive symptoms in PD patients [35]. The expression of c-Abl is increased in trigeminal neuralgia, where it is accompanied by the loss of dopamine neurons in the striatum via aminoacyl-tRNA synthetase-interacting multifunctional protein type2 (AIMP2, p38) activation [28]. In PD models, c-Abl inhibitors reduce phosphorylation of Cdk5, decrease phosphorylation and clearance of α-syn and parkin, and decrease levels of several parkin substrates such as zinc finger protein 746 (PARIS), AIMP2, fuse-binding protein 1 (FBP1), and synphilin-1 [17][32][36][17,32,36]. Radotinib has even been demonstrated to protect primary cortical neurons from toxic cell death induced by c-Abl activation with preformed α-syn fibrils and to reduce Lewy bodies/Lewy neurites-like pathology [35]. Overall, increased activation of c-Abl through parkin inactivation, accumulation of its toxic substrate AIMP2, α-syn aggregation, and impaired autophagy are shown to be associated with neurodegenerative processes of PD.
In AD, c-Abl plays a role in the development of Tau pathology by regulating cytoskeletal signaling cascades. Immunocytochemical studies show that c-Abl is associated with both neuritic plaques and neurofibrillary tangles in the brains of patients with AD. c-Abl interacts directly with Tau and phosphorylates it at tyrosine 394 [23][36][23,36], which has a regulatory effect on normal Tau-related processes, including microtubule assembly and axonal transport, and to trigger aggregation of Tau into paired helical filaments [26]. Neuronal spine pathology is associated with the early onset of AD. Amyloid beta oligomers (AβOs) are known to induce synaptotoxicity, leading to synaptic dysfunction/loss and the reduction in dendritic spine density that underlies cognitive defects [4]. c-Abl was activated in neurons exposed to AβOs and in the brains of patients with AD. Inhibition of active c-Abl ameliorated all AβOs-induced synaptic changes [37] and cognitive deficits in the AD mouse model [4]. AβOs induction of c-Abl signaling appears to involve the tyrosine kinase ephrin receptor A4 (EphA4) [4] and decreases the number of mushroom spines in c-Abl knockout neurons, while preserving the populations of immature stubby, filopodia spines, suggesting that c-Abl deficiency increases the population of immature spines and decreases AβOs-induced synapse elimination [37].
Table 1. c-Abl expression and activity in disease.
| Status | Detected in Patients and Cell or Animal Models | Reference | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Increased c-Abl expression |
|
[11]][11[15],15[23][24,23,24] [38][39][38,39] [9][40][9,40] [9] [41] [23][31][23,31]
|