Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yuyang Jiang | -- | 4678 | 2023-08-24 11:34:31 | | | |
| 2 | Catherine Yang | + 3 word(s) | 4681 | 2023-08-25 03:47:30 | | | | |
| 3 | Catherine Yang | Meta information modification | 4681 | 2023-08-28 09:58:44 | | | | |
| 4 | Catherine Yang | Meta information modification | 4681 | 2023-08-30 04:13:33 | | | | |
| 5 | Catherine Yang | Meta information modification | 4681 | 2023-08-30 04:16:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dai, Q.; Sun, Q.; Ouyang, X.; Liu, J.; Jin, L.; Liu, A.; He, B.; Fan, T.; Jiang, Y. The Antitumor Activity of s-Triazine Derivatives. Encyclopedia. Available online: https://encyclopedia.pub/entry/48426 (accessed on 20 July 2026).
Dai Q, Sun Q, Ouyang X, Liu J, Jin L, Liu A, et al. The Antitumor Activity of s-Triazine Derivatives. Encyclopedia. Available at: https://encyclopedia.pub/entry/48426. Accessed July 20, 2026.
Dai, Qiuzi, Qinsheng Sun, Xiaorong Ouyang, Jinyang Liu, Liye Jin, Ahao Liu, Binsheng He, Tingting Fan, Yuyang Jiang. "The Antitumor Activity of s-Triazine Derivatives" Encyclopedia, https://encyclopedia.pub/entry/48426 (accessed July 20, 2026).
Dai, Q., Sun, Q., Ouyang, X., Liu, J., Jin, L., Liu, A., He, B., Fan, T., & Jiang, Y. (2023, August 24). The Antitumor Activity of s-Triazine Derivatives. In Encyclopedia. https://encyclopedia.pub/entry/48426
Dai, Qiuzi, et al. "The Antitumor Activity of s-Triazine Derivatives." Encyclopedia. Web. 24 August, 2023.
Copy Citation
1,3,5-triazine derivatives, also called s-triazines, are a series of containing-nitrogen heterocyclic compounds that play an important role in anticancer drug design and development. Three s-triazine derivatives, including altretamine, gedatolisib, and enasidenib, have already been approved for refractory ovarian cancer, metastatic breast cancer, and leukemia therapy, respectively, demonstrating that the s-triazine core is a useful scaffold for the discovery of novel anticancer drugs.
s-triazines
antitumor
drug target
1. Topoisomerases Inhibition
In recent years, a number of DNA topoisomerase inhibitors have been reported in both basic research and clinical applications. Topoisomerase was first discovered in E. coli by Wang et al. in 1971. Since then, the study of topoisomerase’s structure, function, and mechanism has been initiated. Generally, human DNA topoisomerases are classified into two large groups: type I and type II topoisomerases. Type I topoisomerases could generate transient single-strand breaks (SSBs) in the DNA molecule by forming a covalent phosphotyrosyl linkage without ATP. In contrast, type II topoisomerases are large homodimeric proteins that require ATP for overall catalytic activity, which could generate transient DNA double-strand breaks (DSBs) [1]. Thus, topoisomerases play important roles in several cell processes, such as replication, transcription, chromosome separation, and segregation. Therefore, topoisomerases are ideal targets for the development of antitumor drugs.
Human DNA topoisomerase inhibitors could be classified into two categories based on their inhibition modes: topoisomerase poisons and topoisomerase catalytic inhibitors [2]. Topoisomerase poisons cover the majority of clinical antitumor agents (e.g., etoposide, doxorubicin, mitoxantrone, salvicine, and teniposide), which can kill cancer cells by stabilizing the covalent topo-DNA complexes and transforming this enzyme into a cellular toxin. Topoisomerase catalytic inhibitors can kill tumor cells by inhibiting the essential enzymatic activity of topoisomerase. A number of structurally diverse compounds have been identified as catalytic inhibitors: preventing DNA cleavage (e.g., merbarone), blocking the enzyme on the ATP-binding site (e.g., purine analogues), or inhibiting ATP hydrolysis (e.g., bisdioxipiperazine analogues). Because of the ubiquitous role of topoisomerase in correlation with various carcinomas, intensive efforts attempting to explore effective tumor therapeutics have led to a panel of chemical agents with diversified structures. In this research, the researchers focused on s-triazine derivatives as topoisomerase inhibitors.
The Novartis research group reported a kind of ATP-competitive catalytic topoisomerase II inhibitor 1 with the core 9H-purine scaffold in 2009 [3], as shown in Figure 1. Then the 9H-purine motif of the compounds was monocyclically substituted by 1,3,5-triazines, which was first reported [4] by Perdih et al. using a virtual screening protocol. In the hit selection, 4-amino-6-(phenylamino)-1,3,5-triazines 2 displayed potent topoisomerase IIa inhibitory activity with IC50 values in the micromolar range (IC50 = 229 µM). Moreover, compound 2 exhibited cytotoxicity against the HepG2 cell lines (IC50 = 20.53 µM), MCF-7 (IC50 = 129.0 µM), and normal HUVEC cells (IC50 = 122 µM). Furthermore, Perdih et al. designed a series of 4,6-disubstituted-1,3,5-triazin-2(1H)-one analogs [5]. This research provided valuable structure-activity relationship (SAR) data regarding the role of substituents introduced at position 6 of the 1,3,5-triazin-2(1H)-one core. Among which, 6-(benzylthio)-4-((3-chlorobenzyl)thio)-1,3,5-triazin-2(1H)-one 3 was identified as a topoisomerase II inhibitor with an improved IC50 of 57.6 µM. However, these compounds showed low cytotoxicity in cancer cells. Perdih et al. further optimize the substituents at position 4 of the 1,3,5-triazin-2(1H)-one scaffold to improve the inhibition potency that would display activity on the cellular level. After virtual screening and experimental evaluation for human topo IIa inhibition, the most potent compounds were 4 and 5, with IC50 values of 8.1 µM and 11.1 µM, respectively, as well as being superior to the positive drug etoposide (IC50 = 28.6 µM) [6]. Additionally, compound 5 showed better HepG2 cell inhibitory activity with EC50 values of 38.7 µM.
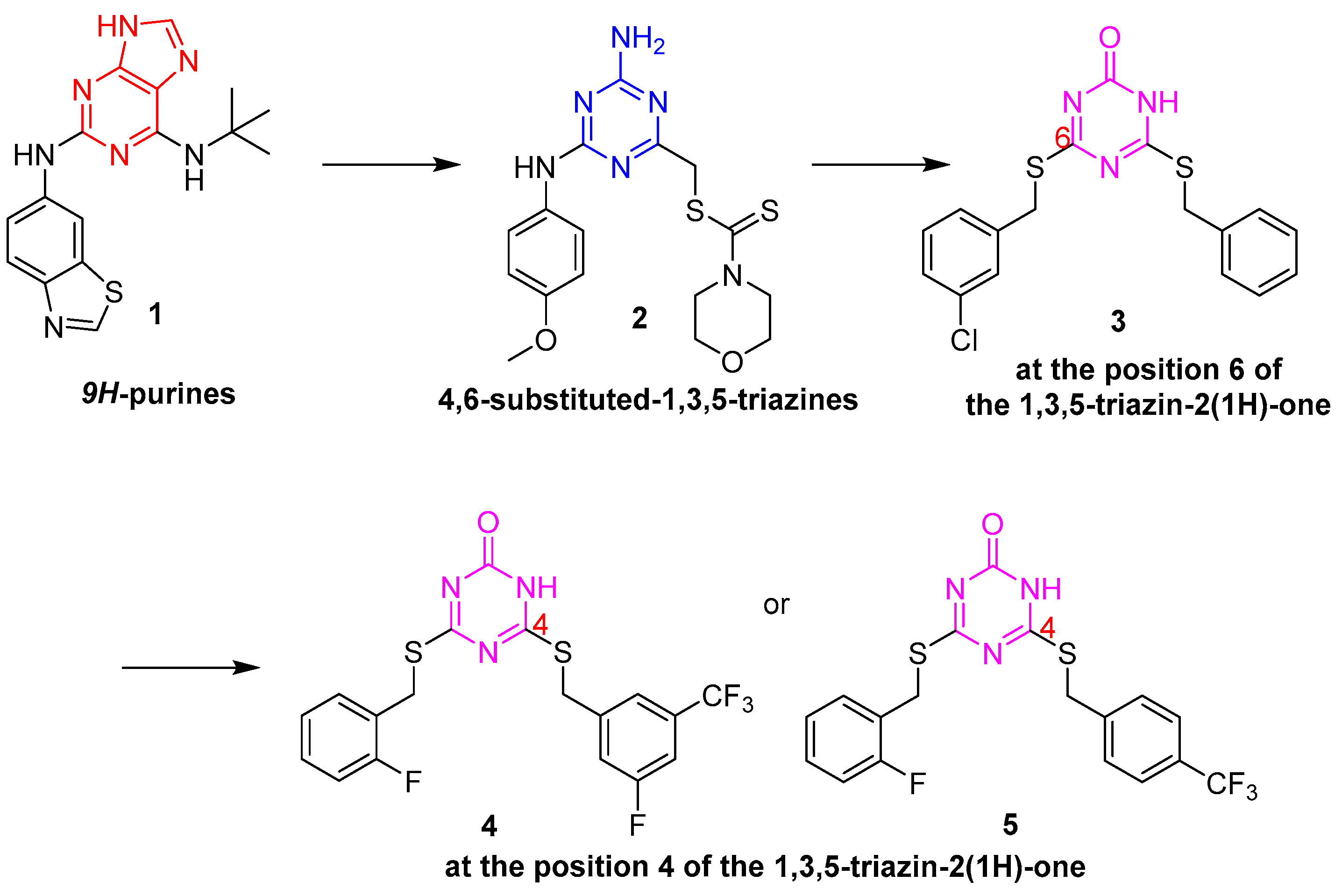
Figure 1. Chemical structures of topoisomerase II inhibitors.
2. Tyrosine Kinases Inhibition
Tyrosine kinases are implicated in tumorigenesis and progression and have emerged as major targets for drug discovery. Tyrosine kinase inhibitors (TKIs) inhibit corresponding kinases from phosphorylating tyrosine residues of their substrates and then block the activation of downstream signaling pathways. Over the past several decades, multiple robust and well-tolerated TKIs with single or multiple targets, including BTK, EGFR, FAK, VEGFR, ALK, ROS1, HER2, MET, MEK, FGFR, and PDGFR, have been developed.
2.1. BTK-TKIs
Bruton’s tyrosine kinase (BTK) is the first Tec family tyrosine kinase and also a key member of the B-cell receptor signalling pathway [7]. Since BTK was confirmed to play a crucial role in B-cell maturation as well as in mast cell activation through the high-affinity IgE receptor, studies of BTK as a target have attracted substantial attention from drug researchers, resulting in the development of a diverse array of BTK inhibitors. BTK inhibitors can be divided into reversible and irreversible inhibitors. Most published BTK inhibitors are irreversible inhibitors, including imidazopyrimidine, 2,4-diaminopyrimidines, imidazoquinoxaline, 1,3,5-trazine, and other groups. There are five covalent irreversible BTK inhibitors (Ibrutinib, Acalabrutinib, Zanubrutinib, Tirabrutinib, Orelabrutinib) that have been marketed so far. Irreversible BTK inhibitors usually react with the key amino acid residue Cys481 of BTK via covalent bond modification. However, they may also inhibit other kinases with structurally related cysteines (such as EGFR, BMX, ITK, and JAK3), resulting in off-target effects and potential toxicity after prolonged administration. Meanwhile, several potent noncovalent reversible inhibitors have also been reported, including ARQ-531, Fenebrutinib, Vecabrutinib, and BIIB068, but none of these reversible BTK inhibitors has yet been approved to date.
Xiang et al. designed a kind of novel covalent BTK inhibitor by substituting the 1H-pyrazolo[3,4-d] pyrimidine core (Ibrutinib) with a 1,3,5-trazines scaffold containing a key acrylamide warhead, as shown in Figure 2. As reported, compound 6 showed potent BTK inhibitory activity (BTK IC50 = 21 nM), validating the potential of the 1,3,5-trazine scaffold as a new skeleton for developing effective BTK inhibitors [8]. However, 6 also potently inhibited EGFR with an IC50 value of 31 nM. In order to improve its selectivity for BTK over EGFR, the 1,3,5-triazine scaffold modified by more hydrophobic side chains, which would additionally occupy the H3 pocket of BTK, was designed. Structure-activity relationship (SAR) and extensive pharmacological screening led to the discovery of a potent irreversible BTK inhibitor 7 [9]. Compound 7 possessed desirable BTK selectivity (IC50 = 17.0 nM) over EGFR (IC50 > 1000 nM) and JAK3 (IC50 = 104.6 nM), which shared a high degree of homology with BTK. However, it is worth noting that broad kinase panel screening is further needed to clarify the selectivity profile of 7.

Figure 2. Chemical structures of BTK inhibitors.
2.2. EGFR-TKIs
The epidermal growth factor receptor (EGFR) is a trans-membrane protein belonging to the erbB/HER family of tyrosine kinase (TK) receptors. EGFR signalling in tumour cells, as opposed to normal cells, is changed and often becomes dysregulated. Accordingly, interrupting EGFR communicating signals is considered one of the targets for curing tumors. Multiple EGFR suppressors have been developed, including 1,3,5-triazine derivatives.
Based on virtual screening, Bai et al. identified a number of 1,3,5-triazine derivatives as potent dual-effective inhibitors against both wild-type and mutant EGFR in 2012 [10]. As shown in Figure 3, compound 8 exhibited the most potent activity against wild-type EGFR (IC50 = 25.9 µM) and mutant EGFR T790M/L858R (IC50 = 6.5 µM). Structure–activity relationships (SAR) were analyzed among the triazine analogs. Compound 9 was obtained by removing the fluorine group in the phenyl part along with the inclusion of the para-hydroxy group on another phenyl, with a low inhibitory potency for wild-type EGFR (IC50 > 100 µM) and reduced about fivefold for mutant EGFR T790M/L858R (IC50 = 30.7 µM). Singh groups reported a novel hybrid analogue of monastrol-1,3,5-triazine 10 as an effective anti-breast cancer agent [11] in 2017, which showed excellent EGFR-TK enzyme inhibitory activity (96.3% at 10 µM). Additionally, their group further modified 1,3,5-triazine to obtain 4-aminoquinoline-1,3,5-triazine derivatives [12]. Compound 11 showed the most EGFR-TK enzyme inhibitory activity (96.4% at 10 µM) and excellent anticancer activities against the entire tested cell lines. In 2021, hybrid quinazoline-1,3,5-triazine derivatives as EGFR inhibitors were reported by Pathak [13]. Through pharmacological evaluation, they found that the substitution of the 1,3,5-triazine ring by morpholine and aniline rings could increase the ligand’s potency in EGFR inhibition. Among them, the most active compound 12 showed an IC50 value of 36.8 nM against the EGFR enzyme.
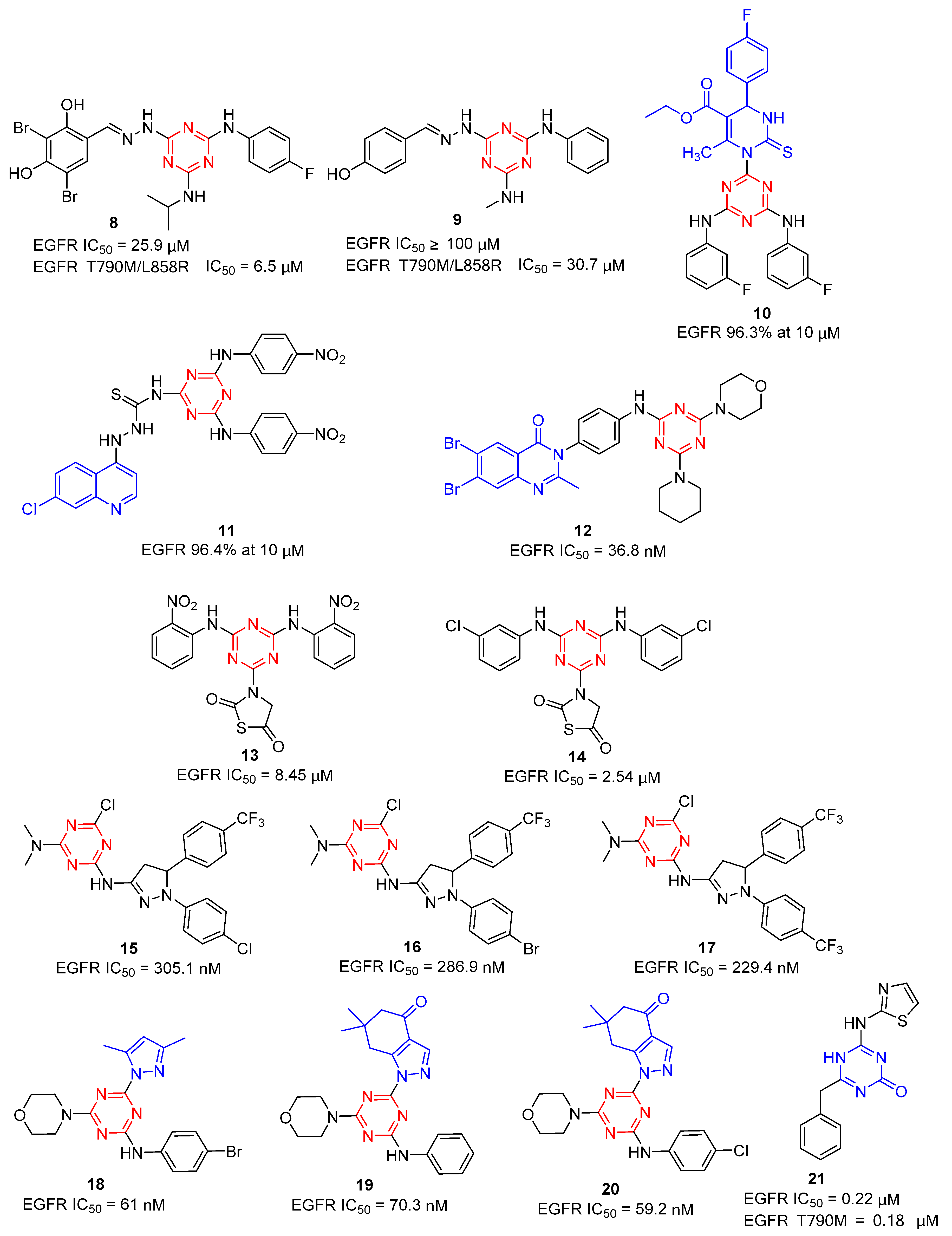
Figure 3. Chemical structures of EGFR inhibitors.
In 2018, He et al. investigated four previously reported 1,3,5-triazine compounds as anti-breast cancer agents via modulation of EGFR-TK [14]. Among the compounds, 13 (8.45 ± 0.65 µM) and 14 (2.54 ± 0.22 µM) significantly inhibited EGFR-TK and exhibited potent anticancer activity in all the tested cell lines in a dose-dependent manner. In 2021, 1,3,5-triazine-based pyrazole derivatives were synthesized by Osman et al. with anticancer activity targeting EGFR [15]. Compounds 15, 16, and 17 displayed excellent activity with an IC50 value of 305.1, 286.9, and 229.4 nM, respectively, in comparison with reference erlotinib.
In 2022, El-Faham et al. synthesized a new series of mono- and bis(dimethylpyrazolyl)-s-triazine derivatives [16]. Of these compounds, 18 showed excellent potency against the HCT116 cell lines (IC50 = 500 ± 80 nM) through apoptosis induction and exhibited potent EGFR inhibitory activity with an IC50 value of 61 nM (tamoxifen, IC50 = 69 nM). For the downstream pathway of PI3K/AKT/mTOR, 18 showed remarkable inhibitory activity with a 0.18-, 0.27-, and 0.39-fold decrease in their concentration. Additionally, pyrazole and fused pyrazole-s-triazine derivatives 19 and 20 were obtained by a one-pot synthesis method, which displayed EGFR inhibitory activity with IC50 values of 70.3 and 59.24 nM, respectively, and similar PI3K/AKT/mTOR inhibitory activity compared with 18 [17]. Therefore, compounds 18, 19, and 20 possessed EGFR/PI3K/AKT/mTOR signaling cascade inhibitors.
EGFR signaling dysregulation in tumors could result in EGFR overexpression or a gain-of-function mutation. According to the reports, acquired drug resistance to the first-generation EGFR-TKIs was revealed due to the T790M and L858R mutations in EGFR. Although a drug related to EGFRT790M or EGFRT790M/L858R mutation has been discovered, it is still needed to discover new EGFR inhibitors with high selectivity for EGFRT790M and EGFRT790M/L858R kinases. Azmy et al. reported a series of 1,3,5-triazine-based derivatives as potential EGFRWT and EGFRT790M inhibitors [18]. Among the derivatives, compound 21 as the most potent antiproliferative agent showed inhibitory activity with EGFRWT (0.22 ± 0.05 µM) and EGFRT790M (0.18 ± 0.11 µM), compared to the reference drugs erlotinib (0.09 ± 0.05 µM) and AZD9291 (0.55 ± 0.10 µM).
2.3. FAK-TKIs
Focal Adhesion Kinase (FAK) is an intracellular non-receptor tyrosine kinase that plays a crucial role in reproduction, early embryonic development, and tumorigenesis through its kinase activity and scaffold function. So far, s-triazines have been proven to be effective inhibitors targeting FAK [19].
In 2013, Chen and co-workers reported a series of diarylamino-1,3,5-triazine derivatives as FAK inhibitors [20]. As shown in Figure 4, compound 22 showed poor activity against FAK (IC50 = 41.9 µM) owing to the introduction of a 3,4,5-trimethoxyanilino group and a methanesulfonamide phenyl group on the 1,3,5-triazine ring. Then, they removed the chlorine atom from 22, and compound 23 with good inhibitory activity against FAK (IC50 = 5.1 µM) was obtained. Moreover, they found that when the methanesulfonamide phenyl group was displaced with an amide moiety, compound 24 showed significantly increased inhibitory activity against FAK (IC50 = 0.4 µM). While changes in the 3,4,5-trimethoxyanilino group of compounds 24 and 25 exhibited similar inhibitory activity against FAK (IC50 = 0.31 µM), in order to improve the FAK kinase activity, they continued to design imidazo[1,2-a][1,3,5] triazines as novel FAK inhibitors in 2015 [21]. The new compounds displayed 10–100 nM IC50 values against FAK, and compound 26 (IC50 = 50 nM) was identified as the best inhibitor.

Figure 4. Chemical structures of FAK inhibitors.
3. Phosphoinositide 3-Kinase Inhibition
Phosphatidylinositol 3-kinase (PI3K) is a lipid kinase, that is, a central component in the PI3K/Akt/mTOR signaling pathway [22]. The most widely studied type I PI3Ks are further subdivided into class IA (PI3Kα, β, and δ) and class IB (PI3Kγ) based on the type of catalytic structural domain. Blocking the PI3K/AKT/mTOR pathway has been widely recognized as an attractive cancer therapeutic strategy owing to its crucial role in cell growth and survival. A number of PI3K inhibitors are currently under research, including pan-PI3K-mTOR inhibitors, pan-class I PI3K inhibitors, and isoformspecific PI3K inhibitors [23]. Dimorpholino-substituted s-triazine derivatives have shown great potential in PI3K-related cancer therapy. Several promising candidates, such as ZSTK474, gedatolisib, and bimiralisib, are currently in clinical trials.
ZSTK474 is an s-triazine derivative that was selected by Zenyaku Kogyo together with more than 1500 other analogues [24]. ZSTK474 potently inhibits the four isoforms of PI3K, with IC50 values of 16, 44, 5, and 49 nM for PI3Kα, β, δ, and γ, respectively, suggesting that it is a pan-PI3K inhibitor [25]. Unfortunately, ZSTK-474 has been withdrawn from clinical trials due to its resistance and on-target/off-tumor side effects [26]. Therefore, the structure of ZSTK-474 needs to be optimized to improve its selective targeting of PI3K.
As shown in Figure 5, D. Ross et al., using ZSTK474 as a lead compound, explored compound 27 by displacing a single morpholine group in ZSTK474 with piperazine. Compound 27 showed a 36-fold reduction in its PI3Kα (IC50 = 180 nM) and PI3Kδ (IC50 = 142 nM) inhibition and a >70-fold reduction in PI3Kβ (IC50 = 1093 nM) and PI3Kγ (IC50 = 1873 nM) inhibition, indicating the sensitivity of this region towards oxygen replacement [27]. However, N-acetylation of 27 to provide 28 showed PI3K isoform inhibition (IC50 = 2.9−21 nM) similar to ZSTK474 (IC50 = 3.9−20.8 nM). To further investigate the role of the morpholine oxygen in these compounds on PI3K inhibition, they found that when the 2-aminoethyl functional groups were displaced with a single morpholine group of ZSTK474 [28], bifunctional inhibitors 29 and 30 displayed nanomolar inhibition towards PI3Kα (IC50 = 130 nM and 107 nM, respectively) and PI3Kδ (IC50 = 236 nM and 137 nM, respectively) and low micromolar inhibition for PI3Kβ and PI3Kγ (IC50 = 1.5−3.9 µM) in enzymatic inhibition assays. Cell viability assays demonstrated that compound 29 showed superior anti-proliferative activities over compound 30 in three tumor-derived cell lines (A375, D54, and SET-2), owing to 29 being able to inhibit downstream AKT and ERK1/2 phosphorylation.
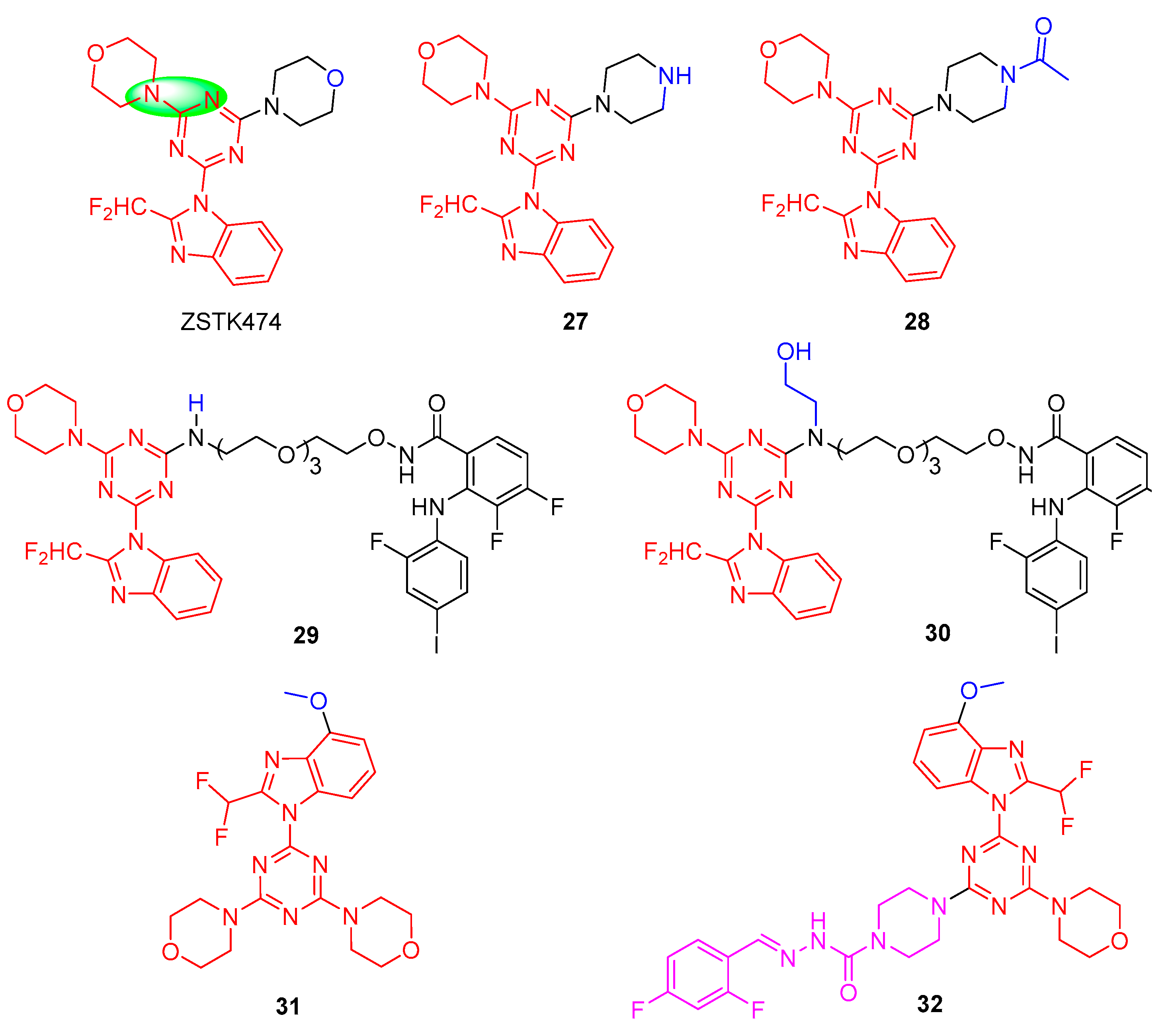
Figure 5. Chemical structures of PI3K inhibitors of ZSTK474 and its derivatives.
In 2011, Shepherd and co-workers found that the introduction of a methoxy group (31) at the 4′-position of the 2-(difluoromethyl)-1H-benzo[d]imidazol of ZST474 could enhance interaction with the PI3Kα protein [29]. Based on this, Hou et al. designed a new 1,3,5-triazine derivative 32 containing semicarbazones as new potential PI3Kα inhibitors, 32 also further by replacing a single morpholine group of 31 with piperazine [30]. Further research indicated that compound 32 displayed excellent inhibitory activity with an IC50 value of 0.32 nM against PI3Kα. Meanwhile, in the U87-MG human glioblastoma xenograft model assay by intragastric administration, 32 exhibited similar antitumor activity at 20 mg/kg/day compared with ZSTK-474 at 40 mg/kg/day in vivo antitumor efficacy. However, the pharmacokinetic properties of 32 are not ideal, so the compound needs to be further modified structurally to improve its physicochemical properties.
Gedatolisib (PKI-587), another dimorpholino-substituted s-triazine derivative developed by Pfizer, is in a phase III clinical trial as a potential agent for the treatment of acute myeloid leukemia, hormone receptor-positive breast cancer, and HER2-negative breast cancer. Gedatolisib exhibits potent in an in vitro inhibitory activity against both PI3K and mTOR and displays significant antitumor effects in vivo xenograft model. However, it suffers from low selectivity over different PI3K isozymes, leading to possible off-target effects and reducing its therapeutic utility. In addition, gedatolisib must be administered by injection, which may not be suitable as a practical drug for cancer patients. Hence, structural modification of gedatolisib is desirable to develop PI3Ks and mTOR inhibitors with enhanced isozyme selectivity and metabolic stability.
The Venkatesan group has reported the bis-morpholino s-triazine-based compounds as potent dual PI3K/mTOR inhibitors [31]. Gedatolisib was found to have poor plasma levels when administered orally, which may owe to its poor permeability, low clogP (1.24), and high molecular weight (Mw: 615). In order to increase the clogP value and reduce molecular weight, Mansour et al. reported a series of mono-morpholino 1,3,5-triazine derivatives by a single morpholine group in gedatolisib that were replaced with 3-oxa-8-azabicyclo[3.2.1]octane [32]. As shown in Figure 6, 33 showed significantly increased potency against PI3Kα (IC50 = 8 nM) and mTOR (IC50 = 0.42 nM) and excellent cellular potency against both MDA-361 (IC50 = 22 nM) and PC3mm2 (IC50 = 29 nM) cell lines. Pharmacokinetic studies of 33 showed good oral bioavailability in nude mice after oral administration (10 mg/kg) and a high half-life (>60 min). Metabolite evaluation studies found that the ethylene bridge on the bridged-morpholine group was the main site of metabolism, and its metabolite structure was determined to be 34 [33]. Therefore, compound 33 is an orally effective dual PI3K/mTOR inhibitor.
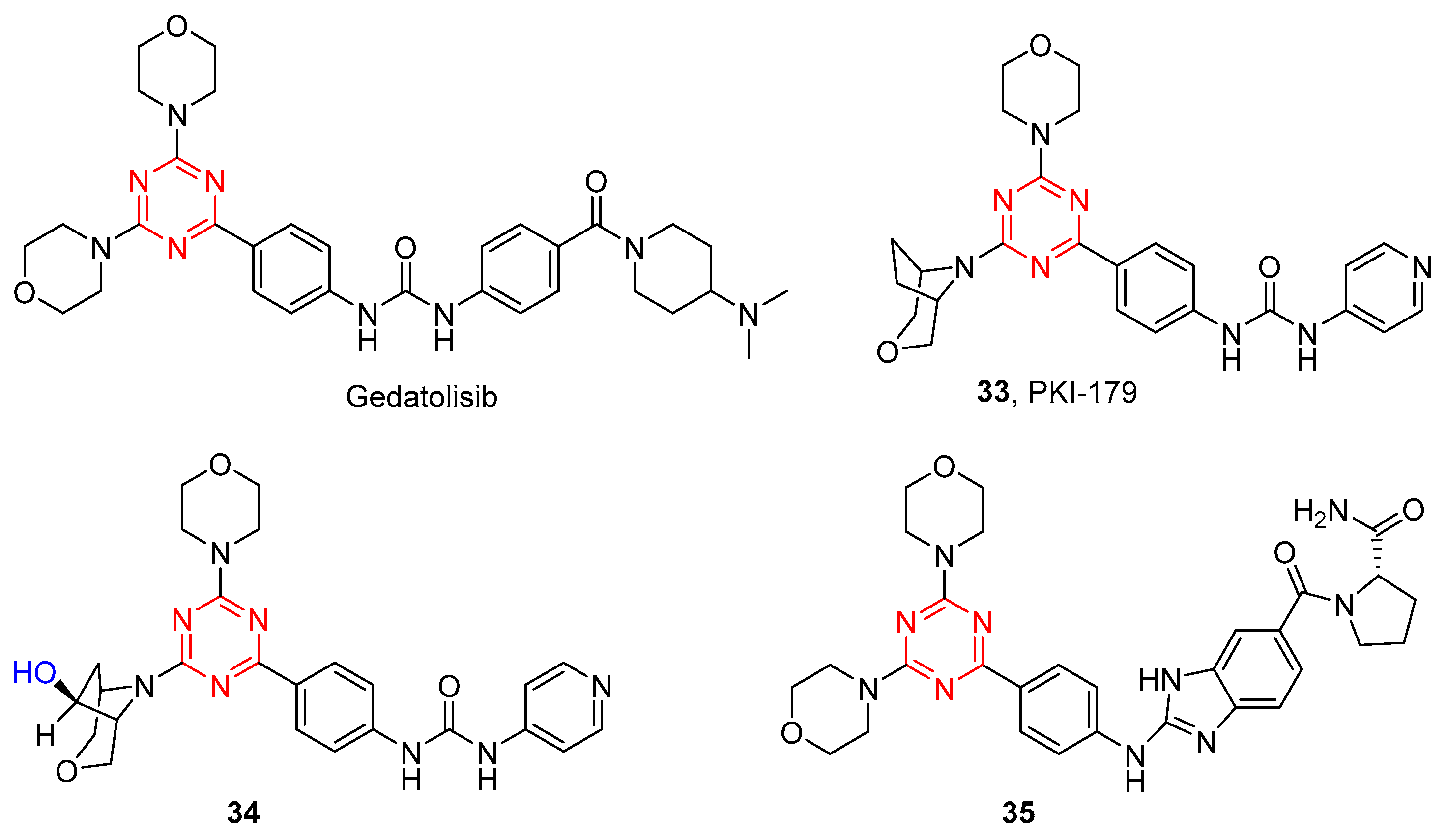
Figure 6. Chemical structures of PI3K inhibitors of gedatolisib and its derivatives.
Zhang et al. designed and synthesized a series of novel substituted s-triazines bearing a benzimidazole group as PI3K/mTOR inhibitors using the ring closing and scaffold hopping methods [34]. Compound 35 showed remarkable inhibitory activity against PI3Kδ with an IC50 value of 2.3 nM and moderate activity against other class I PI3K isoforms and mTOR (with IC50 values of 14.6, 34.0, 849.0, and 15.4 nM for PI3Kα, β, γ, and mTOR, respectively). Therefore, compound 35 could significantly suppress the PI3K/Akt/mTOR signaling pathway.
BKM120 is one of the clinically most advanced pan-PI3K inhibitors, but it interferes with an off-target effect on microtubule polymerization. As shown in Figure 7, Wymann et al. have identified that it differs from BKM120 by having only one atom that could be separated into discrete PI3K and tubulin activities [35]. Initially inspired by ZSTK474 and BKM120, they replaced the BKM120 core with s-triazine to yield bimiralisib (36), aiming to maximize compound solubility and bioavailability and avoid microtubule interactions. As expected, 36 displayed potent inhibition against pan-PI3K and showed no microtubule-destabilizing agent activity [36]. Above all, 36 is a highly selective pan-PI3K inhibitor with balanced targeting of mTOR kinase, which passed phase I studies and is now in phase II studies in relapsed and refractory lymphoma and advanced solid tumors [37][38].

Figure 7. Chemical structures of PI3K inhibitors of BKM120 and its derivatives.
Subsequently, Wymann and co-workers mainly focused on the 4-(difluoromethyl)-pyrimidin-2-amine group as an optimized residue for PI3K binding; compound 37 was obtained by substituting 4-(trifluoromethyl)-pyridine-2-amine of 36 with the group. Compound 37 showed low nanomolar affinity PI3Kα (IC50 = 2.2 nM) and demonstrated significant antitumor activity in a mice xenograft model at a concentration almost eight times lower than the parental phase-II inhibitor 35 [39]. Recently, starting from the lead compound 37, they designed various potent covalent PI3Kα inhibitors by targeting the solvent-exposed cysteines at a distance > 10 Å from an ATP-site-directed core group [40]. A number of compounds bearing an acrylamide warhead and different linker modules were synthesized to assess the required warhead reactivity and the spatial trajectory of the Michael acceptor. Among these compounds, compound 38 showed the most PI3Kα enzyme inhibitory activity (IC50 = 1 nM) and excellent stability in rat liver microsomes.
Based on the structures of PI3K inhibitor BKM120 and Hh inhibitor vismodegib, the researchers' group reported a novel series of unsymmetrical diaryl ureas as potent inhibitors simultaneously inhibiting PI3K/Akt/mTOR and Hh signalings [41]. After biological activities were examined, compound 39 showed excellent antiproliferative activity against MDA-MB-231, T47D, and MCF-7 cells. The enzymatic activity of 39 (IC50 = 180 nM) against mTOR was of nanomolecular value. Furthermore, based on the structures of the PI3K inhibitor ZSTK474 and the CRBN ligand pomalidomide, the researchers have designed a series of new small-molecule PROTACs for the degradation of PI3K [42]. Compound 40 indicated great potency on PI3Kα with an IC50 of 24 nM and could inhibit tumor cell proliferation by inducing autophagy instead of apoptosis or cell cycle arrest.
In 2012, Baselga and Wulf’s groups independently found that PI3K inhibition could promote HR deficiency by downregulating BRCA1/2 and could sensitize BRCA-proficient tumors to PARP inhibition, which provides an effective therapy for the combined administration of PI3K and PARP inhibitors to expand the application of PARP inhibitors [43][44]. Based on these works, Xu et al. reported a series of dual PARP/PI3K inhibitors by merging the pharmacophores of PARP and PI3K inhibitors [45]. As shown in Figure 8, structure−activity relationships (SARs) indicate that the R1 group, which serves as a hydrogen-bond donor, could be modified to change the compounds’ PI3K inhibitory activity. Several compounds were synthesized and evaluated for PARP-1 and PI3Kα inhibitory activities. As expected, all the remaining compounds showed excellent PARP-1 inhibitory activity in the low nanomolar range. The first round of structural optimization provided compound 41, which displayed appropriate PARP-1 and PI3Kα inhibitory activities with IC50 values of 9.08 and 7.04 nM, respectively. Subsequently, to further optimize the 1,3,5-triazine core by a bicyclic system of tetrahydropyrido[3,4-d]pyrimidine, compound 42 displayed the most promising candidate with IC50 values of 8.22 and 8.25 nM against PARP-1 and PI3Kα. Next, they replaced the phthalazine moiety with a benzofuran-7-carboxamide moiety to obtain compound 43, which exhibited good PARP-1 and PI3Ka inhibitory activity with an inhibition ratio of more than 50% at 10 nM and 100 nM, respectively. After SAR screening and experimental evaluation, the most potent compound was 44 against PARP-1 and PI3Kα with IC50 values of 13.8 and 64.0 nM, respectively [46].
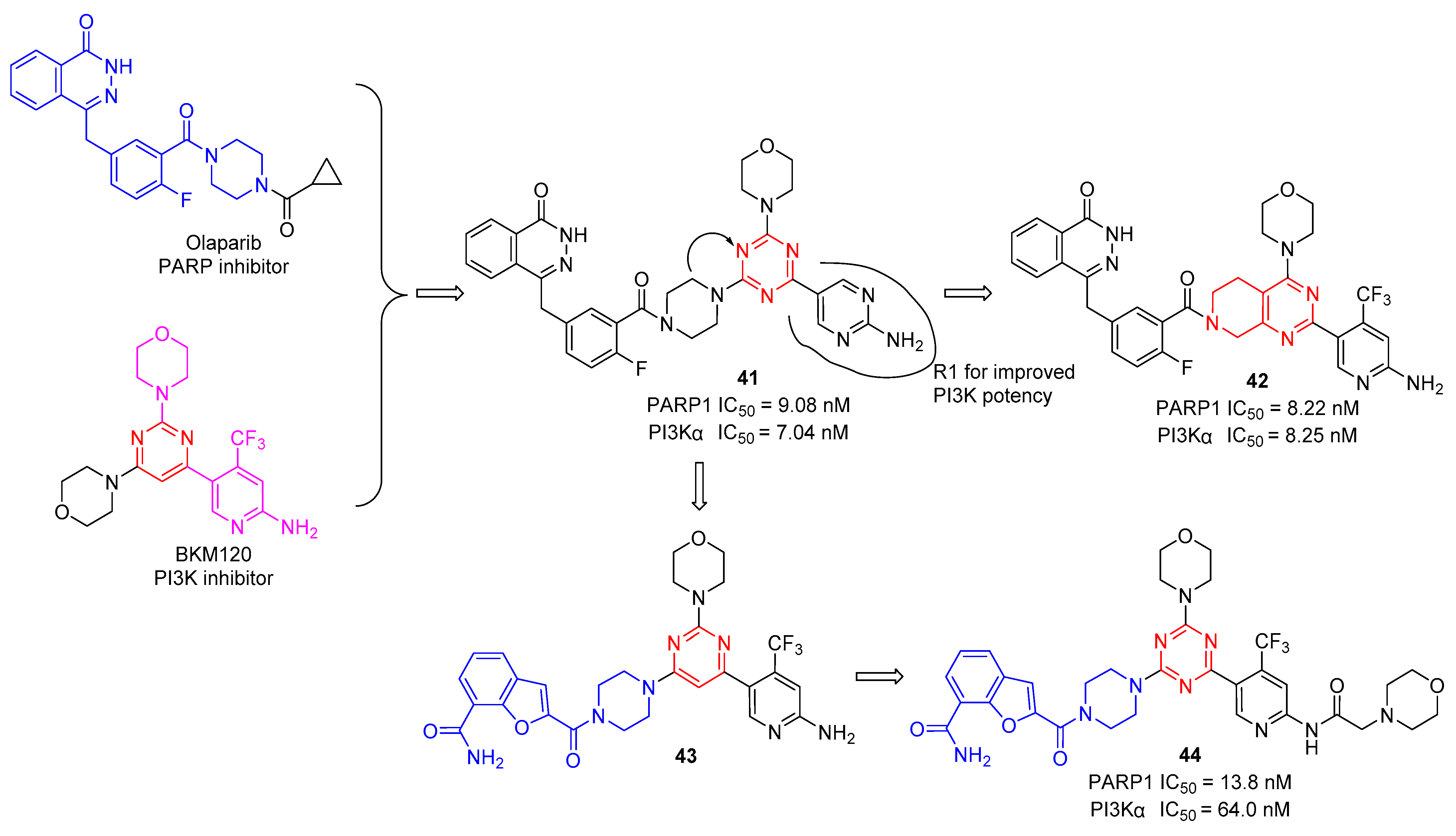
Figure 8. Chemical structures of s-triazine simultaneous targeting PI3K and PARP.
The Ras/MEK/ERK and PI3K/Akt/mTOR pathways play important biological functions. MEK inhibition could promote a compensatory activation of PI3K/Akt kinase activity. As shown in Figure 9, Ross et al. reported a s-triazine analog 45 by combining the PI3K inhibitor ZSTK474 with the Raf/MEK inhibitor RO5126766. Bifunctional inhibitor 45 showed good inhibitory activity against MEK1 (IC50 = 473 nM) and PI3K (IC50 = 172 nM) [47]. However, the corresponding inhibition of pERK1/2 activity was low (35−40%) in two representative human cancer cell lines (A549, PANC-1) at the 5 μM concentration level. Recently, they continued to report a series of bifunctional MEK1/PI3K inhibitors by covalent linking the PI3K inhibitor ZSTK474 and the MEK inhibitor PD0325901 [48]. Among them, compound 46 showed excellent inhibition against MEK1 (IC50 = 0.015 nM) and PI3K (IC50 = 191 nM) and displayed a 95% and 67% inhibition of tumor ERK1/2 and Akt phosphorylation, respectively, at 2 h postadministration.
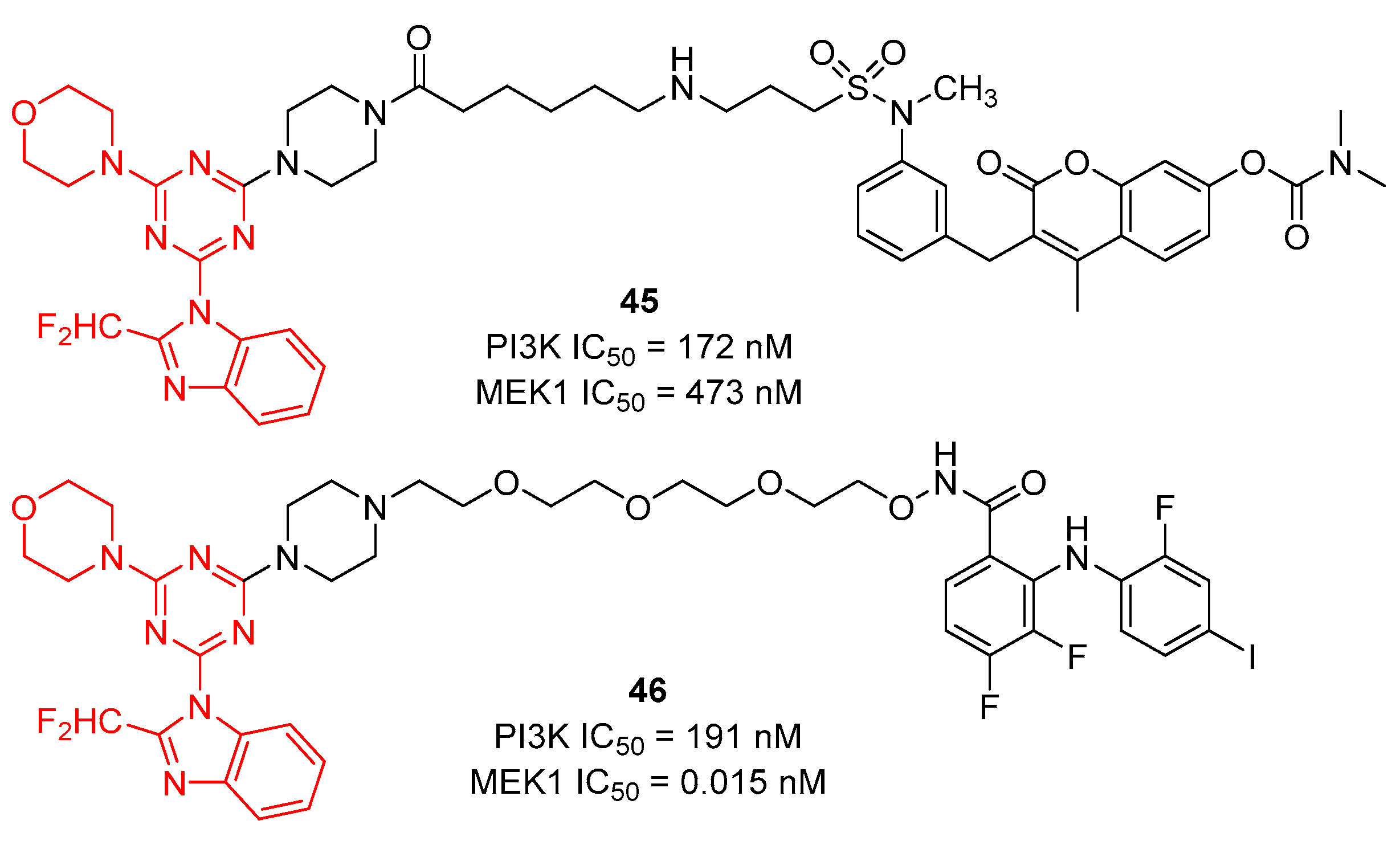
Figure 9. Chemical structures of PI3K and MEK bifunctional inhibitors.
Zhu and co-workers reported 2-(thiophen-2-yl)-1,3,5-triazine derivatives 47 as PI3K and mTOR inhibitors in 2020 [49]. By experimental screening, as shown in Figure 10, compound 47 showed excellent inhibition activity of cell proliferation against A549, MCF-7, and Hela cancer cell lines with IC50 values of 0.20 ± 0.05 µM, 1.25 ± 0.11 µM, and 1.03 ± 0.24 µM, and potently inhibited PI3K and mTOR kinases with IC50 values of 7.0 and 48 nM, respectively. Further, they also reported the 2-arylurea-1,3,5-triazine derivative 48 as a PI3K and mTOR inhibitor [50]. Compound 48 exhibited potent inhibition against PI3K and mTOR kinases with IC50 values of 23.8 and 10.9 nM, respectively. Compound 48 was further evaluated in MCF-7 cells and MCF-7 xenograft models, which showed significant in vitro and in vivo anticancer efficacies. Besides, in order to enhance the mTOR kinase activity, they introduced aryl urea units with various substituents into the triazine core and modified it with arylurea by pyridine to obtain a series of novel thiophene-triazine derivatives bearing arylurea [51]. Among them, the inhibitory activity of 49 against PI3Kα and mTOR kinase was excellent, with IC50 values of 177.41 and 12.24 nM, respectively, indicating that it was a potential dual PI3Kα/mTOR inhibitor.
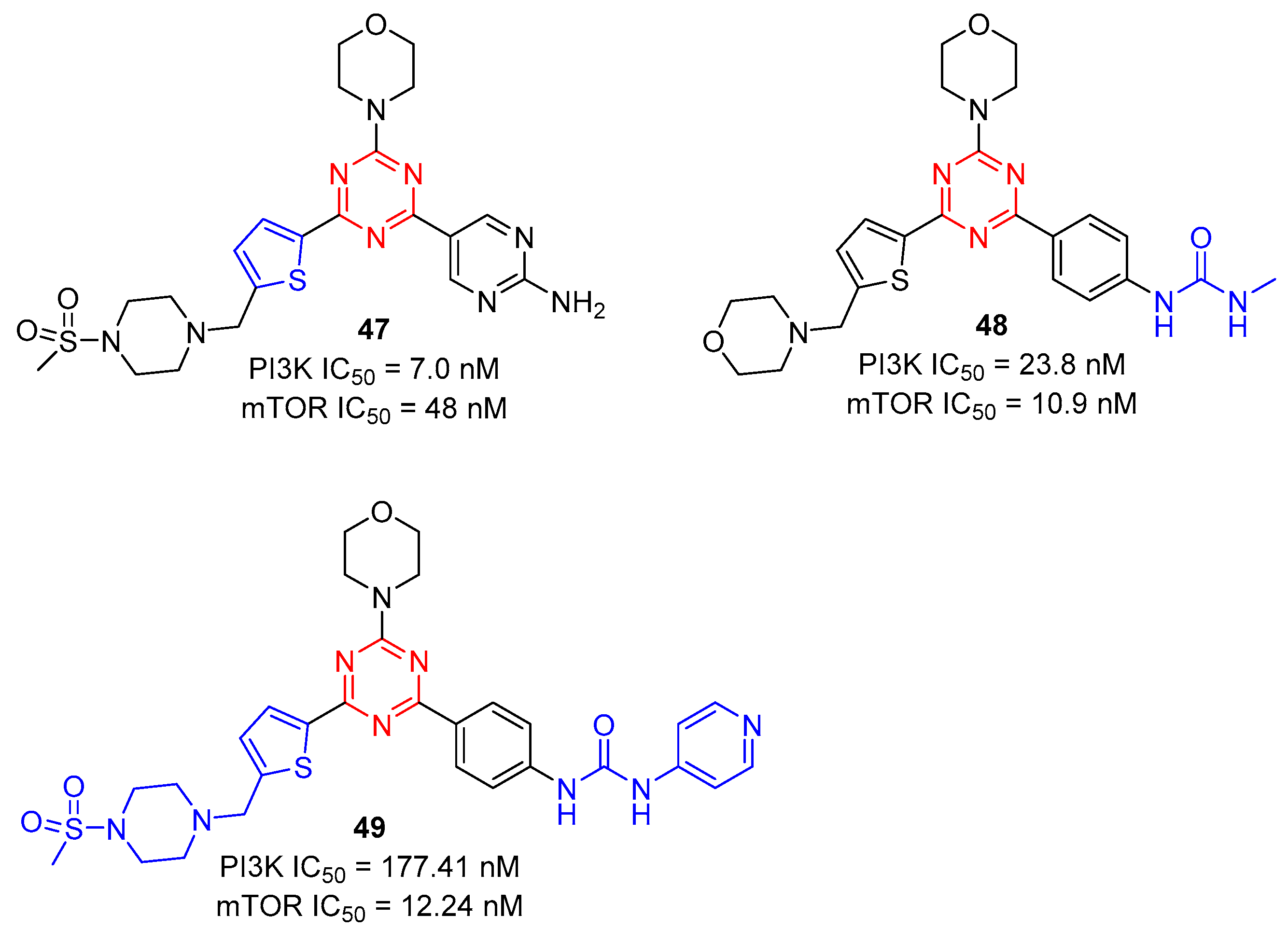
Figure 10. Chemical structures of PI3K and mTOR bifunctional inhibitors.
4. NADP+-Dependent Isocitrate Dehydrogenases Inhibition
Metabolic reprogramming is a hallmark of cancer, promoting the initiation and maintenance of tumors. The NADP+-dependent isocitrate dehydrogenases (IDH) are critical metabolic enzymes, converting isocitrate to α-ketoglutarate (αKG) in the tricarboxylic acid cycle. IDH mutations occur in multiple tumors, mainly in IDH1 (R132) and IDH2 (R140 and R172). In recent years, several IDH inhibitors have been approved by the FDA. Among them, AG-221 (enasidenib) is a first-in-class, orally available, small-molecule s-triazine IDH2 inhibitor [52]. Through high-throughput screening, as shown in Figure 11, Su et al. [53] reported several s-triazine compounds as effective inhibitors against IDH2R140Q, and the initial hit compound 50 was obtained. Compound 50 showed micromolar inhibitory potency for IDH2R140Q with IC50 values of 1.9 µM. SAR analysis among the s-triazine analogs obtained compound 51, the first nanomolar inhibitor of IDH2R140Q (IC50 = 7 nM at 16 h). Although compound 51 showed potency in enzymatic and cellular assays, 51 displayed high lipophilicity, resulting in solubility-limited absorption in vivo. Additionally, it emerged poor in vitro liver microsomal stability, which translated to high clearance in vivo. Thus, they continue optimizing the substituents around the s-triazine core. Through the addition of mildly polar substituents with trifluoromethyl pyridine and 2-methyl-2-propanol to provide 52, AG-221(52) showed excellent potency for 2HG inhibition, improved solubility, low clearance, and good oral bioavailability in vivo in rats, supporting the clinical trials of AG-221 in patients with IDH2 mutation.
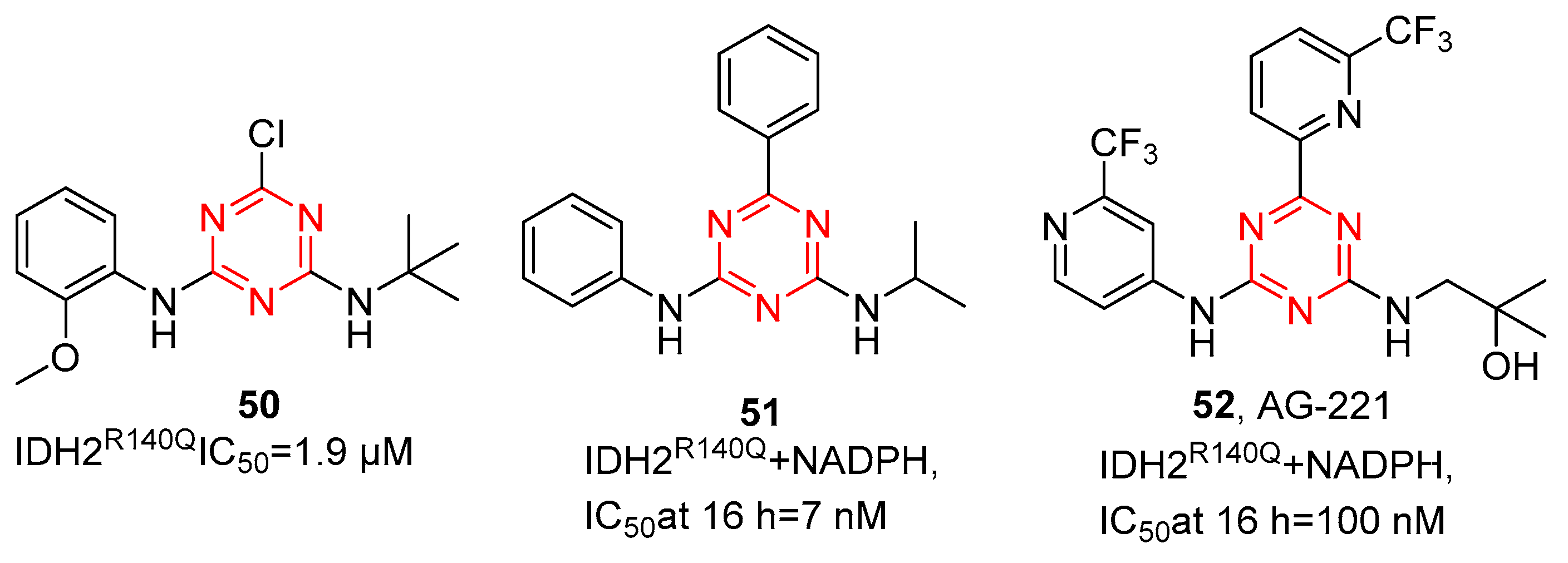
Figure 11. Chemical structures of IDH inhibitors of AG-221 and its derivatives.
5. Cyclin-Dependent Kinases Inhibiton
Cyclin-dependent kinases (CDKs) are members of a complex family of heterodimeric serine/threonine protein kinases that play an important role in regulating cell cycle machinery. Depending on their biological functions, CDKs can be divided into cell cycle regulatory CDKs (CDK1, CDK2, CDK4, and CDK6 are included) and transcription-associated CDKs (CDK7−9, CDK11−13, and CDK19 are included) [54]. Murray et al. have identified [1,3,5]triazine-pyridine as a new series of potent CDK inhibitors [55]. Among them, compound 53 (Figure 12) displayed excellent inhibitory potency at CDK1 (IC50 = 0.021 µM), CDK2 (IC50 = 0.007 µM), and CDK5 (IC50 = 0.003 µM) and submicromolar potency at CDK4 (IC50 = 0.308 µM), CDK6 (IC50 = 0.356 µM), and CDK7 (IC50 = 0.126 µM). The broad spectrum of potent CDK inhibitory activities and the in vitro and in vivo antitumor efficacy of 53 may render it as a valuable pharmacological tool in elucidating the complex roles of CDK signaling pathways.
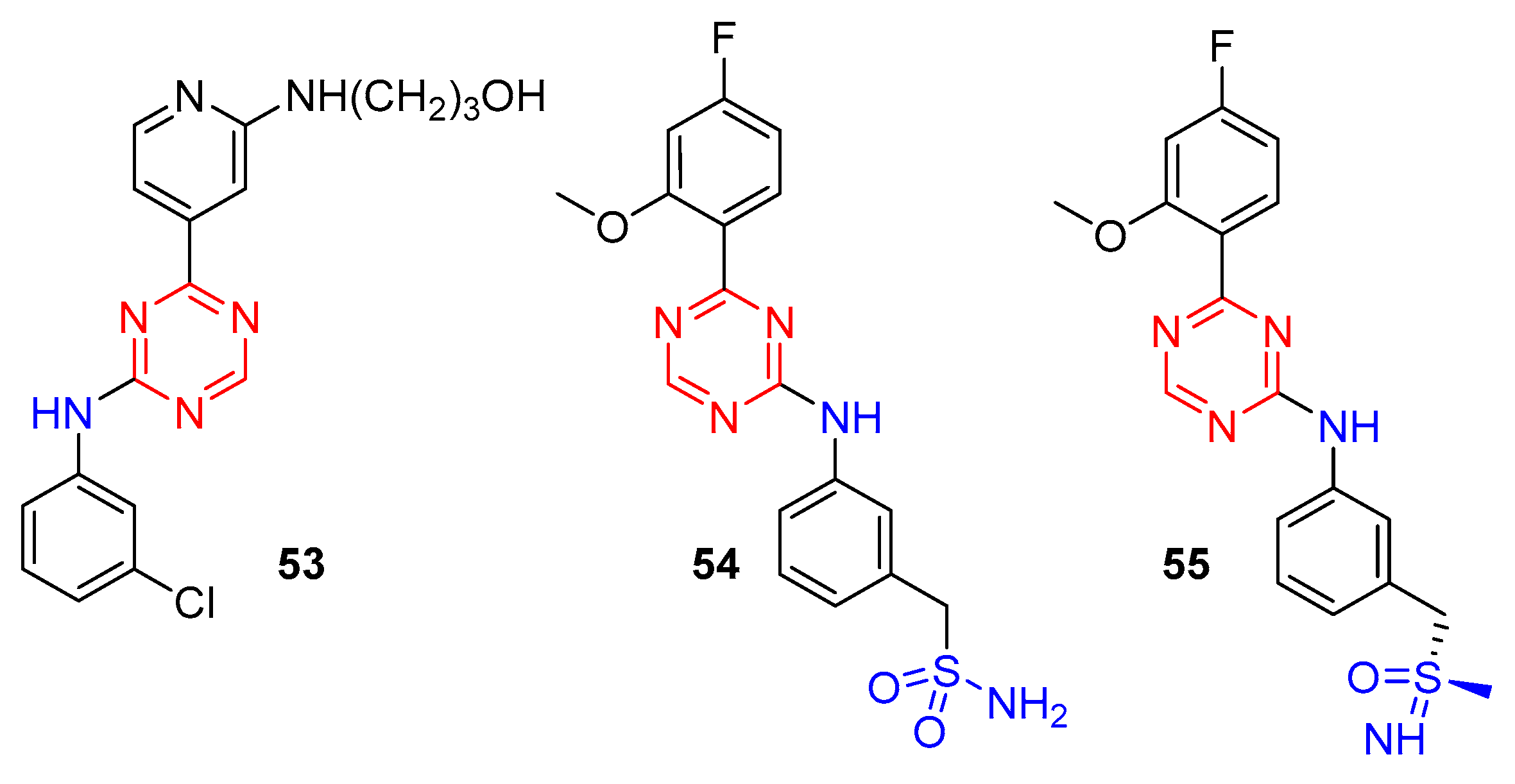
Figure 12. Chemical structures of s-triazine targeting CDK.
Positive transcription elongation factor b (PTEFb) is a hetero-dimer of CDK9 and one of four cyclin partners: cyclin T1, cyclin K, cyclin T2a, or cyclin T2b. Deregulated kinase activity of CDK9 of the PTEFb hetero-dimer is associated with cancer. Starting from lead compound 54, Lucking et al. have optimized structurally characterized by an unuaual benzyl sulfoximine group to obtain 55 [56]. Through kinase selectivity, physicochemical properties, DMPK properties, in vitro and in vivo efficacy, 55 is the first potent and highly selective PTEFb/CDK9 (IC50 CDK9/CycT1: 13 nm, ratio of IC50 values CDK2/CDK9: 100) inhibitor to enter clinical trials for the treatment of cancer.
6. Others
Human adenosine receptors (hARs) can be classified into four subtypes: hA1, hA2A, hA2B, and hA3. All four belong to the G protein-coupled receptor (GPCR) family, and each has a different pharmacological profile, tissue distribution, and function. Langmead et al., in an effort to find a novel hit by virtual screening, reported several compounds containing 1,3,5-triazine that bind to hA1 and hA2A. Subsequently, 1,3,5-triazine analogs were identified as potent human adenosine receptor antagonists for Parkinson’s disease [57][58]. To modify s-triazine to produce selective ligands for subtypes other than hA2A. Yu et al. designed and synthesized novel 1,3,5-triazine derivatives targeting the hA1 and hA3 adenosine receptors for cancer therapy [59]. As shown in Figure 13, compounds 56 and 57 showed good binding affinity to both hA1 (IC50 = 139.3 nM, 78.1 nM, respectively) and hA3 AR (IC50 = 55.5 nM, 13.3 nM, respectively) and could inhibit cell viability, leading to cell death in lung cancer cell lines.

Figure 13. Chemical structures of hAR inhibitors 56, 57, Hsp90 inhibitors 58, PD-L1. Inhibitors 59, and MMP and VEGF inhibitors 60.
Heat shock protein 90 (Hsp90), a kind of molecular chaperone, is widely expressed and highly conserved in cells. DCZ5248 (58, Figure 13), a triazine derivative by the Lou group, is a novel Hsp90 inhibitor that directly binds to the Hsp90 protein and does not induce a heat shock response [60], and identified 58 was a dual inhibitor of both Hsp90 and late-autophagy with potent antitumor activity against colon cancer cells in vitro and in vivo.
Programmed cell death protein 1 (PD-1) and its ligand PD-L1 comprise immune checkpoints located on T-cells. The PD-1/PD-L1 axis is hijacked by tumor cells to suppress immune surveillance. At present, inhibition of the PD-1/PD-L1 axis by monoclonal antibodies has achieved remarkable success and is approved for cancer. However, a number of small molecules targeting PD-L1 have been reported [61]. Marinelli and co-workers have identified 2,4,6-tri- and 2,4-disubstituted 1,3,5-triazines as PD-L1 inhibitors [62]. Among them, compound 59 demonstrated to strongly bind with the PD-L1 protein with a nanomolar IC50 of 0.115 µM and not to PD-1, as shown in Figure 13.
Matrix metalloproteinases (MMPs) are a characteristic family of zinc-dependent endopeptidases that could promote extracellular matrix turnover, tumor growth, angiogenesis, and metastasis. Considering MMP-9 could promote cell migration and trigger the angiogenic switch during carcinogenesis via expression of vascular endothelial growth factor (VEGF), therefore, Khattab et al. developed a series of star-shaped triazine-based dendrimers as MMP and VEGF inhibitors [63]. As shown in Figure 13, the most active anticancer dendrimer 60 showed potent MMP-9, -10, and -13 inhibitors with IC50 values of 156, 145, and 124 nM, respectively. Furthermore, 60 suppressed the correlated oncogenic mediators, VEGF expression, induced apoptosis (>75%), and inhibited tumor cell migration (∼84%) in Caco-2 cells.
References
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2003, 88, 118–122.
- Hu, W.; Huang, X.-S.; Wu, J.-F.; Yang, L.; Zheng, Y.-T.; Shen, Y.-M.; Li, Z.-Y.; Li, X. Discovery of novel topoisomerase II inhibitors by medicinal chemistry approaches. J. Med. Chem. 2018, 61, 8947–8980.
- Furet, P.; Schoepfer, J.; Radimerski, T.; Chene, P. Discovery of a new class of catalytic topoisomerase II inhibitors targeting the ATP-binding site by structure based design. Part I. Bioorg. Med. Chem. Lett. 2009, 19, 4014–4017.
- Pogorelcnik, B.; Brvar, M.; Zajc, I.; Filipic, M.; Solmajer, T.; Perdih, A. Monocyclic 4-amino-6-(phenylamino)-1,3,5-triazines as inhibitors of human DNA topoisomerase II alpha. Bioorg. Med. Chem. Lett. 2014, 24, 5762–5768.
- Pogorelcnik, B.; Janezic, M.; Sosic, I.; Gobec, S.; Solmajer, T.; Perdih, A. 4,6-Substituted-1,3,5-triazin-2(H-1)-ones as monocyclic catalytic inhibitors of human DNA topoisomerase II alpha targeting the ATP binding site. Bioorg. Med. Chem. 2015, 23, 4218–4229.
- Bergant, K.; Janezic, M.; Valjavec, K.; Sosic, I.; Pajk, S.; Stampar, M.; Zegura, B.; Gobec, S.; Filipic, M.; Perdih, A. Structure-guided optimization of 4,6-substituted-1,3,5-triazin-2(1H)-ones as catalytic inhibitors of human DNA topoisomerase II alpha. Eur. J. Med. Chem. 2019, 175, 330–348.
- Liang, C.Y.; Tian, D.N.; Ren, X.D.; Ding, S.J.; Jia, M.Y.; Xin, M.H.; Thareja, S. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: A mini-review. Eur. J. Med. Chem. 2018, 151, 315–326.
- Teng, Y.; Lu, X.; Xiao, M.X.; Li, Z.B.; Zou, Y.M.; Ren, S.N.; Cheng, Y.; Luo, G.S.; Xiang, H. Discovery of potent and highly selective covalent inhibitors of Bruton’s tyrosine kinase bearing triazine scaffold. Eur. J. Med. Chem. 2020, 199, 112339.
- Xiao, M.; Zhu, M.; Wu, S.; Ma, L.; Qi, L.; Ha, S.; Xiong, S.; Chen, M.; Chen, D.; Luo, G.; et al. Novel 6-amino-1,3,5-triazine derivatives as potent BTK inhibitors: Structure-activity relationship (SAR) analysis and preliminary mechanism investigation. Bioorg. Chem. 2023, 130, 106263.
- Bai, F.; Liu, H.; Tong, L.; Zhou, W.; Liu, L.; Zhao, Z.; Liu, X.; Jiang, H.; Wang, X.; Xie, H.; et al. Discovery of novel selective inhibitors for EGFR-T790M/L858R. Bioorg. Med. Chem. Lett. 2012, 22, 1365–1370.
- Srivastava, J.K.; Pillai, G.G.; Bhat, H.R.; Verma, A.; Singh, U.P. Design and discovery of novel monastrol-1,3,5-triazines as potent anti-breast cancer agent via attenuating Epidermal Growth Factor Receptor tyrosine kinase. Sci. Rep. 2017, 7, 1038–1045.
- Bhat, H.R.; Singh, U.P.; Thakur, A.; Ghosh, S.K.; Gogoi, K.; Prakash, A.; Singh, R.K. Design, synthesis, anticancer, antibacterial, and antifungal evaluation of 4-aminoquinoline-1,3,5-triazine derivatives. J. Heterocycl. Chem. 2019, 57, 390–399.
- Pathak, P.; Rimac, H.; Grishina, M.; Verma, A.; Potemkin, V. Hybrid quinazoline 1,3,5-triazines as Epidermal Growth Factor Receptor (EGFR) Inhibitors with anticancer activity: Design, synthesis, and computational study. ChemMedChem 2021, 16, 822–838.
- Yan, W.; Zhao, Y.; He, J. Anti-breast cancer activity of selected 1,3,5-triazines via modulation of EGFR-TK. Mol. Med. Rep. 2018, 18, 4175–4184.
- Raghu, M.S.; Pradeep Kumar, C.B.; Prashanth, M.K.; Yogesh Kumar, K.; Prathibha, B.S.; Kanthimathi, G.; Alissa, S.A.; Alghulikah, H.A.; Osman, S.M. Novel 1,3,5-triazine-based pyrazole derivatives as potential antitumor agents and EFGR kinase inhibitors: Synthesis, cytotoxicity, DNA binding, molecular docking and DFT studies. New J. Chem. 2021, 45, 13909–13924.
- Shawish, I.; Barakat, A.; Aldalbahi, A.; Malebari, A.M.; Nafie, M.S.; Bekhit, A.A.; Albohy, A.; Khan, A.; Ul-Haq, Z.; Haukka, M.; et al. Synthesis and Antiproliferative Activity of a New Series of Mono- and Bis(dimethylpyrazolyl)-s-triazine Derivatives Targeting EGFR/ PI3K/AKT/mTOR Cascades. Acs Omega 2022, 7, 24858–24870.
- Shawish, I.; Barakat, A.; Aldalbahi, A.; Alshaer, W.; Daoud, F.; Alqudah, D.A.; Al Zoubi, M.; Hatmal, M.m.M.; Nafie, M.S.; Haukka, M.; et al. Acetic Acid Mediated for One-Pot Synthesis of Novel Pyrazolyl s-Triazine Derivatives for the Targeted Therapy of Triple-Negative Breast Tumor Cells (MDA-MB-231) via EGFR/PI3K/AKT/mTOR Signaling Cascades. Pharmaceutics 2022, 14, 1558.
- Hashem, H.E.; Amr, A.E.-G.E.; Nossier, E.S.; Anwar, M.M.; Azmy, E.M. New Benzimidazole-, 1,2,4-Triazole-, and 1,3,5-Triazine-Based Derivatives as Potential EGFRWT and EGFRT790M Inhibitors: Microwave-Assisted Synthesis, Anticancer Evaluation, and Molecular Docking Study. Acs Omega 2022, 7, 7155–7171.
- Pang, X.-J.; Liu, X.-J.; Liu, Y.; Liu, W.-B.; Li, Y.-R.; Yu, G.-X.; Tian, X.-Y.; Zhang, Y.-B.; Song, J.; Jin, C.-Y.; et al. Drug Discovery Targeting Focal Adhesion Kinase (FAK) as a Promising Cancer Therapy. Molecules 2021, 26, 4250.
- Dao, P.; Jarray, R.; Le Coq, J.; Lietha, D.; Loukaci, A.; Lepelletier, Y.; Hadj-Slimane, R.; Garbay, C.; Raynaud, F.; Chen, H. Synthesis of novel diarylamino-1,3,5-triazine derivatives as FAK inhibitors with anti-angiogenic activity. Bioorg. Med. Chem. Lett. 2013, 23, 4552–4556.
- Dao, P.; Smith, N.; Tornkiewicz-Raulet, C.; Yen-Pon, E.; Carnacho-Artacho, M.; Lietha, D.; Herbeuval, J.-P.; Commoul, X.; Garbay, C.; Chen, H. Design, Synthesis, and Evaluation of Novel Imidazo 1,2-a 1,3,5 triazines and Their Derivatives as Focal Adhesion Kinase Inhibitors with Antitumor Activity. J. Med. Chem. 2015, 58, 237–251.
- Tong, G.; Peng, T.; Chen, Y.; Sha, L.; Dai, H.; Xiang, Y.; Zou, Z.; He, H.; Wang, S. Effects of GLP-1 Receptor Agonists on Biological Behavior of Colorectal Cancer Cells by Regulating PI3K/AKT/mTOR Signaling Pathway. Front. Pharmacol. 2022, 13, 901559.
- Bertucci, A.; Bertucci, F.; Goncalves, A. Phosphoinositide 3-Kinase (PI3K) Inhibitors and Breast Cancer: An Overview of Current Achievements. Cancers 2023, 15, 1416.
- Yaguchi, S.; Izumisawa, Y.; Sato, M.; Nakagane, T.; Koshimizu, I.; Sakita, K.; Kato, M.; Yoshioka, K.; Sakato, M.; Kawashima, S. In vitro cytotoxicity of imidazolyl-1,3,5-triazine derivatives. Biol. Pharm. Bull. 1997, 20, 698–700.
- Kong, D.; Yamori, T. ZSTK474 is an ATP-competitive inhibitor of class I phosphatidylinositol 3 kinase isoforms. Cancer Sci. 2007, 98, 1638–1642.
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; Andre, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769.
- Van Dort, M.E.; Galban, S.; Nino, C.A.; Hong, H.; Apfelbaum, A.A.; Luker, G.D.; Thurber, G.M.; Atangcho, L.; Besirli, C.G.; Ross, B.D. Structure-Guided Design and Initial Studies of a Bifunctional MEK/PI3K Inhibitor(ST-168). Acs Med. Chem. Lett. 2017, 8, 808–813.
- Van Dort, M.E.; Jang, Y.; Bonham, C.A.; Heist, K.; McDonald, L.; Zhang, E.Z.; Chenevert, T.L.; Luker, G.D.; Ross, B.D. Structural effects of morpholine replacement in ZSTK474 on Class I PI3K isoform inhibition: Development of novel MEK/PI3K bifunctional inhibitors. Eur. J. Med. Chem. 2022, 229, 113996.
- Rewcastle, G.W.; Gamage, S.A.; Flanagan, J.U.; Frederick, R.; Denny, W.A.; Baguley, B.C.; Kestell, P.; Singh, R.; Kendall, J.D.; Marshall, E.S.; et al. Synthesis and Biological Evaluation of Novel Analogues of the Pan Class I Phosphatidylinositol 3-Kinase (PI3K) Inhibitor 2-(Difluoromethyl)-1- 4,6-di(4-morpholinyl)-1,3,5-triazin-2-yl -1H-benzi midazole (ZSTK474). J. Med. Chem. 2011, 54, 7105–7126.
- Wang, Y.; Liu, Y.; Ge, T.; Tang, J.; Wang, S.; Gao, Z.; Chen, J.; Xu, J.; Gong, P.; Zhao, Y.; et al. Based on 2-(difluoromethyl)-1- 4,6-di (4-morpholinyl)-1,3,5-triazin-2-yl -1H-benzimidazole (ZSTK474), design, synthesis and biological evaluation of novel PI3K alpha selective inhibitors. Bioorg. Chem. 2023, 130, 106211.
- Venkatesan, A.M.; Dehnhardt, C.M.; Delos Santos, E.; Chen, Z.; Dos Santos, O.; Ayral-Kaloustian, S.; Khafizova, G.; Brooijmans, N.; Mallon, R.; Hollander, I.; et al. Bis(morpholino-1,3,5-triazine) Derivatives: Potent Adenosine 5′-Triphosphate Competitive Phosphatidylinositol-3-kinase/Mammalian Target of Rapamycin Inhibitors: Discovery of Compound 26 (PKI-587), a Highly Efficacious Dual Inhibitor. J. Med. Chem. 2010, 53, 2636–2645.
- Venkatesan, A.M.; Chen, Z.; Dos Santos, O.; Dehnhardt, C.; Delos Santos, E.; Ayral-Kaloustian, S.; Mallon, R.; Hollander, I.; Feldberg, L.; Lucas, J.; et al. PKI-179: An orally efficacious dual phosphatidylinositol-3-kinase (PI3K)/mammalian target of rapamycin (mTOR) inhibitor. Bioorg. Med. Chem. Lett. 2010, 20, 5869–5873.
- Chen, Z.; Venkatesan, A.M.; Dos Santos, O.; Delos Santos, E.; Dehnhardt, C.M.; Ayral-Kaloustian, S.; Ashcroft, J.; McDonald, L.A.; Mansour, T.S. Stereoselective Synthesis of an Active Metabolite of the Potent PI3 Kinase Inhibitor PKI-179. J. Org. Chem. 2010, 75, 1643–1651.
- Wu, T.-T.; Guo, Q.-Q.; Chen, Z.-L.; Wang, L.-L.; Du, Y.; Chen, R.; Mao, Y.-H.; Yang, S.-G.; Huang, J.; Wang, J.-T.; et al. Design, synthesis and bioevaluation of novel substituted triazines as potential dual PI3K/mTOR inhibitors. Eur. J. Med. Chem. 2020, 204, 112637.
- Bohnacker, T.; Prota, A.E.; Beaufils, F.; Burke, J.E.; Melone, A.; Inglis, A.J.; Rageot, D.; Sele, A.M.; Cmiljanovic, V.; Cmiljanovic, N.; et al. Deconvolution of Buparlisib’s mechanism of action defines specific PI3K and tubulin inhibitors for therapeutic intervention. Nat. Commun. 2017, 8, 14683.
- Beaufils, F.; Cmiljanovic, N.; Cmiljanovic, V.; Bohnacker, T.; Melone, A.; Marone, R.; Jackson, E.; Zhang, X.; Sele, A.; Borsari, C.; et al. 5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-ami ne (PQR309), a Potent, Brain-Penetrant, Orally Bioavailable, Pan-Class I PI3K/mTOR Inhibitor as Clinical Candidate in Oncology. J. Med. Chem. 2017, 60, 7524–7538.
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 Is a Novel Dual PI3K/mTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin. Cancer Res. 2018, 24, 120–129.
- Wicki, A.; Brown, N.; Xyrafas, A.; Bize, V.; Hawle, H.; Berardi, S.; Cmiljanovic, N.; Cmiljanovic, V.; Stumm, M.; Dimitrijevic, S.; et al. First-in human, phase 1, dose-escalation pharmacokinetic and pharmacodynamic study of the oral dual PI3K and mTORC1/2 inhibitor PQR309 in patients with advanced solid tumors (SAKK 67/13). Eur. J. Cancer 2018, 96, 6–16.
- Borsari, C.; Rageot, D.; Beaufils, F.; Bohnacker, T.; Keles, E.; Buslov, I.; Melone, A.; Sele, A.M.; Hebeisen, P.; Fabbro, D.; et al. Preclinical Development of PQR514, a Highly Potent PI3K Inhibitor Bearing a Difluoromethyl-Pyrimidine Moiety. Acs Med. Chem. Lett. 2019, 10, 1473–1479.
- Borsari, C.; Keles, E.; McPhail, J.A.; Schaefer, A.; Sriramaratnam, R.; Goch, W.; Schaefer, T.; De Pascale, M.; Bal, W.; Gstaiger, M.; et al. Covalent Proximity Scanning of a Distal Cysteine to Target PI3K alpha. J. Am. Chem. Soc. 2022, 144, 6326–6342.
- Li, W.; Sun, Q.; Song, L.; Gao, C.; Liu, F.; Chen, Y.; Jiang, Y. Discovery of 1-(3-aryl-4-chloropheny1)-3-(p-aryl)urea derivatives against breast cancer by inhibiting PI3K/Akt/mTOR and Hedgehog signalings. Eur. J. Med. Chem. 2017, 141, 721–733.
- Li, W.; Gao, C.; Zhao, L.; Yuan, Z.; Chen, Y.; Jiang, Y. Phthalimide conjugations for the degradation of oncogenic PI3K. Eur. J. Med. Chem. 2018, 151, 237–247.
- Ibrahim, Y.H.; Garcia-Garcia, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzman, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047.
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmana, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K Inhibitor with a PARP Inhibitor Provides an Effective Therapy for BRCA1-Related Breast Cancer. Cancer Discov. 2012, 2, 1048–1063.
- Wang, J.; Li, H.; He, G.; Chu, Z.; Peng, K.; Ge, Y.; Zhu, Q.; Xu, Y. Discovery of Novel Dual Poly(ADP-ribose)polymerase and Phosphoinositide 3-Kinase Inhibitors as a Promising Strategy for Cancer Therapy. J. Med. Chem. 2020, 63, 122–139.
- Wang, J.; He, G.; Li, H.; Ge, Y.; Wang, S.; Xu, Y.; Zhu, Q. Discovery of novel PARP/PI3K dual inhibitors with high efficiency against BRCA-proficient triple negative breast cancer. Eur. J. Med. Chem. 2021, 213, 113054.
- Van Dort, M.E.; Galban, S.; Wang, H.; Sebolt-Leopold, J.; Whitehead, C.; Hong, H.; Rehemtulla, A.; Ross, B.D. Dual inhibition of allosteric mitogen-activated protein kinase (MEK) and phosphatidylinositol 3-kinase (PI3K) oncogenic targets with a bifunctional inhibitor. Bioorg. Med. Chem. 2015, 23, 1386–1394.
- Van Dort, M.E.; Hong, H.; Wang, H.; Nino, C.A.; Lombardi, R.L.; Blanks, A.E.; Galban, S.; Ross, B.D. Discovery of Bifunctional Oncogenic Target Inhibitors against Allosteric Mitogen-Activated Protein Kinase (MEK1) and Phosphatidylinositol 3-Kinase (PI3K). J. Med. Chem. 2016, 59, 2512–2522.
- Zhang, B.; Zhang, Q.; Xiao, Z.; Sun, X.; Yang, Z.; Gu, Q.; Liu, Z.; Xie, T.; Jin, Q.; Zheng, P.; et al. Design, synthesis and biological evaluation of substituted 2-(thiophen-2-yl)-1,3,5-triazine derivatives as potential dual PI3K alpha/mTOR inhibitors. Bioorg. Chem. 2020, 95, 103525.
- Sun, X.; Zhang, B.; Luo, L.; Yang, Y.; He, B.; Zhang, Q.; Wang, L.; Xu, S.; Zheng, P.; Zhu, W. Design, synthesis and pharmacological evaluation of 2-arylurea-1,3,5-tria-zine derivative (XIN-9): A novel potent dual PI3K/mTOR inhibitor for cancer therapy. Bioorg. Chem. 2022, 129, 106157.
- Xu, S.; Luo, L.; Sun, X.; Yang, Y.; Guo, Q.; Jiang, Z.; Wu, Y. Design, synthesis and antitumor activity of novel thiophene- triazine derivatives bearing arylurea unit as potent PI3K/mTOR inhibitorss. Bioorganic Med. Chem. 2023, 78, 117133.
- Kim, E.S. Enasidenib: First Global Approval. Drugs 2017, 77, 1705–1711.
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493.
- Teng, Y.; Lu, K.; Zhang, Q.; Zhao, L.; Huang, Y.; Ingarra, A.M.; Galons, H.; Li, T.; Cui, S.; Yu, P.; et al. Recent advances in the development of cyclin-dependent kinase 7 inhibitors. Eur. J. Med. Chem. 2019, 183, 111641.
- Kuo, G.H.; DeAngelis, A.; Emanuel, S.; Wang, A.H.; Zhang, Y.; Connolly, P.J.; Chen, X.; Gruninger, R.H.; Rugg, C.; Fuentes-Pesquera, A.; et al. Synthesis and identification of 1,3,5 triazine-pyridine biheteroaryl as a novel series of potent cyclin-dependent kinase inhibitors. J. Med. Chem. 2005, 48, 4535–4546.
- Luecking, U.; Scholz, A.; Lienau, P.; Siemeister, G.; Kosemund, D.; Bohlmann, R.; Briem, H.; Terebesi, I.; Meyer, K.; Prelle, K.; et al. Identification of Atuveciclib (BAY 1143572), the First Highly Selective, Clinical PTEFb/CDK9 Inhibitor for the Treatment of Cancer. ChemMedChem 2017, 12, 1776–1793.
- Masih, A.; Singh, S.; Agnihotri, A.K.; Giri, S.; Shrivastava, J.K.; Pandey, N.; Bhat, H.R.; Singh, U.P. Design and development of 1,3,5-triazine-thiadiazole hybrids as potent adenosine A(2)A receptor (A(2)AR) antagonist for benefit in Parkinson’s disease. Neurosci. Lett. 2020, 735, 135222.
- Masih, A.; Agnihotri, A.K.; Srivastava, J.K.; Pandey, N.; Bhat, H.R.; Singh, U.P. Discovery of novel 1,3,5-triazine as adenosine A(2A) receptor antagonist for benefit in Parkinson’s disease. J. Biochem. Mol. Toxicol. 2021, 35, e22659.
- Park, S.; Ahn, Y.; Kim, Y.; Roh, E.J.; Lee, Y.; Han, C.; Yoo, H.M.; Yu, J. Design, Synthesis and Biological Evaluation of 1,3,5-Triazine Derivatives Targeting hA1 and hA3 Adenosine Receptor. Molecules 2022, 27, 4016.
- Chen, X.-l.; Liu, P.; Zhu, W.-l.; Lou, L.-g. DCZ5248, a novel dual inhibitor of Hsp90 and autophagy, exerts antitumor activity against colon cancer. Acta Pharmacol. Sin. 2021, 42, 132–141.
- Zarganes-Tzitzikas, T.; Konstantinidou, M.; Gao, Y.; Krzemien, D.; Zak, K.; Dubin, G.; Holak, T.A.; Domling, A. Inhibitors of programmed cell death 1 (PD-1): A patent review (2010–2015). Expert Opin. Ther. Pat. 2016, 26, 973–977.
- Russomanno, P.; Assoni, G.; Amato, J.; D’Amore, V.M.; Scaglia, R.; Brancaccio, D.; Pedrini, M.; Polcaro, G.; La Pietra, V.; Orlando, P.; et al. Interfering with the Tumor-Immune Interface: Making Way for Triazine-Based Small Molecules as Novel PD-L1 Inhibitors. J. Med. Chem. 2021, 64, 16020–16045.
- Haiba, N.S.; Khalil, H.H.; Bergas, A.; Abu-Serie, M.M.; Khattab, S.N.; Teleb, M. First-in-Class Star-Shaped Triazine Dendrimers Endowed with MMP-9 Inhibition and VEGF Suppression Capacity: Design, Synthesis, and Anticancer Evaluation. Acs Omega 2022, 7, 21131–21144.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
5 times
(View History)
Update Date:
30 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No