 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | David J. Kennedy | -- | 2323 | 2023-08-22 03:56:07 |
Video Upload Options
Vascular calcification (VC) is one of the major causes of cardiovascular morbidity and mortality in patients with chronic kidney disease (CKD). VC is a complex process expressing similarity to bone metabolism in onset and progression. VC in CKD is promoted by various factors not limited to hyperphosphatemia, Ca/Pi imbalance, uremic toxins, chronic inflammation, oxidative stress, and activation of multiple signaling pathways in different cell types, including vascular smooth muscle cells (VSMCs), macrophages, and endothelial cells.
1. Introduction
|
Promoters |
Inhibitors |
|---|---|
|
Bone morphogenetic protein (BMP) 2, 4, and 6 Osteocalcin Alkaline phosphatase (Sex determining region Y)-box 9 (SOX9) Osterix Matrix metalloproteinase (MMP) 2, 3, and 7 Runt-related transcription factor 2 (Runx2) Calcium Phosphate Glucose Advanced glycation end products Oxidized low-density lipoproteins Collagen I Receptor activator of nuclear factor-kB ligand (RANKL) |
Matrix Gla protein (MGP) Osteopontin Osteoprotegerin Vitamin K Magnesium Bone morphogenetic protein 7 (BMP7) Fetuin Klotho Parathyroid hormone (PTH) Pyrophosphate Carbonic anhydrase Collagen IV Inorganic pyrophosphate (PPi) |
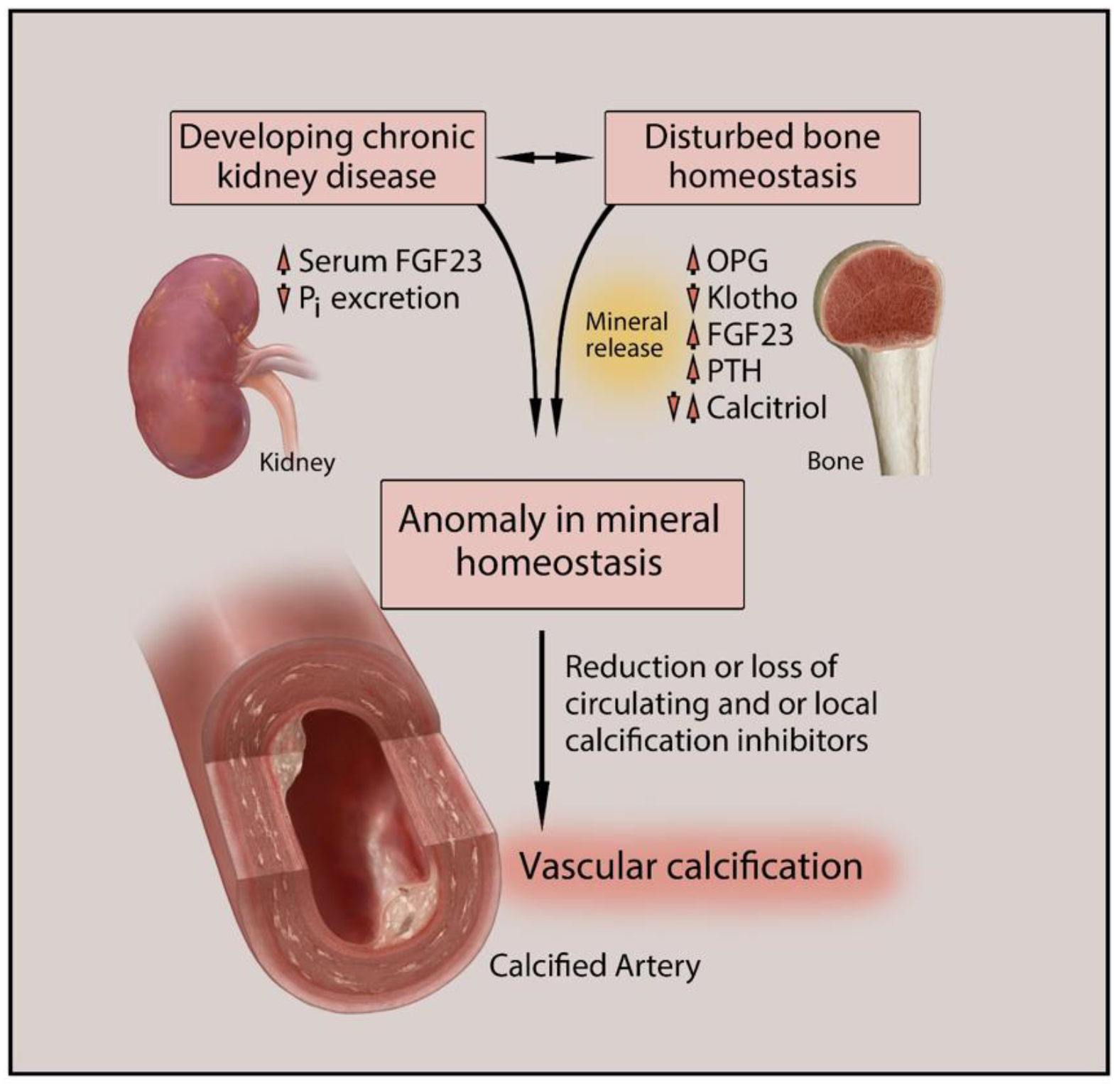
2. Types and Anatomical Presence of VC
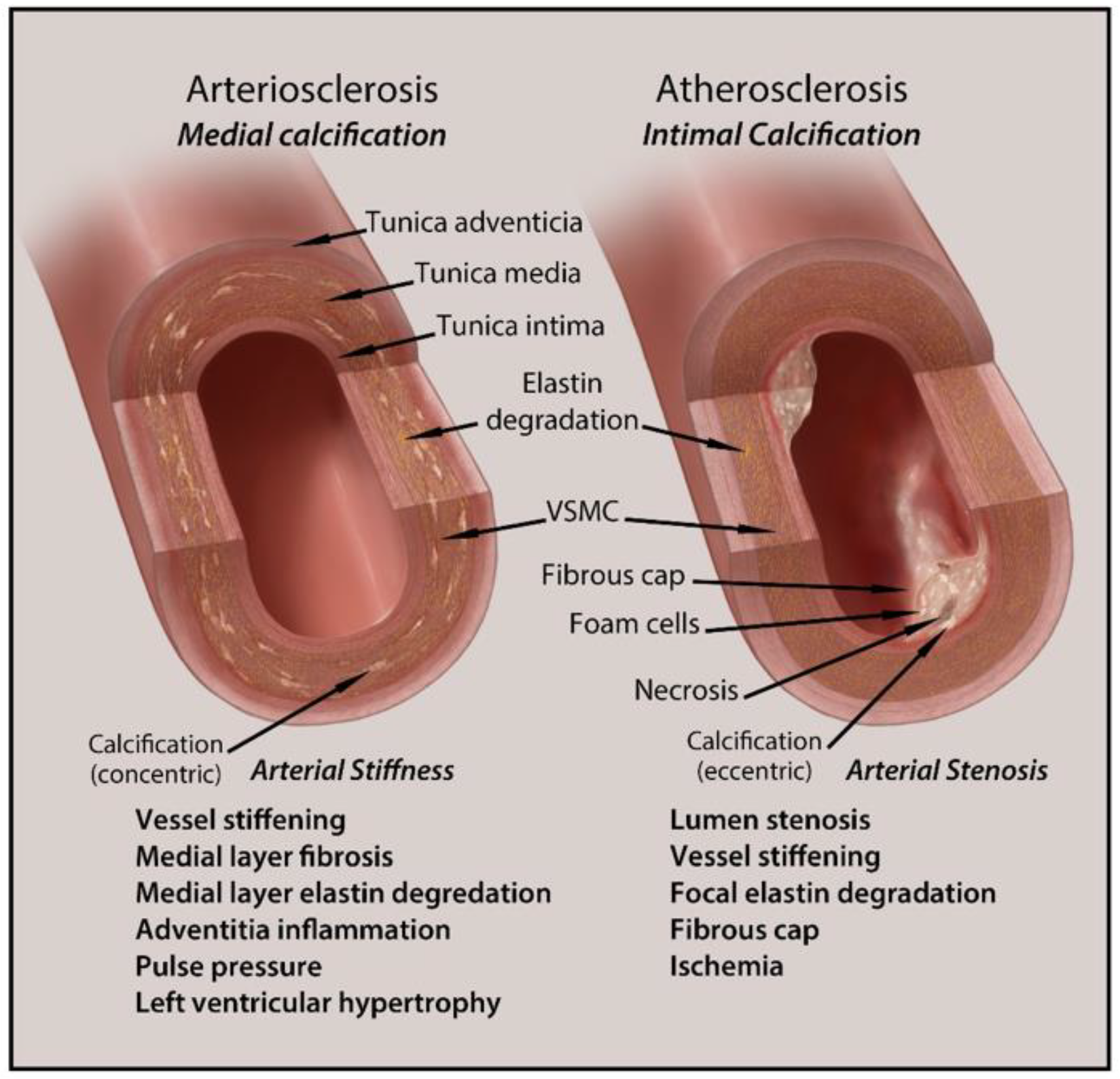
3. Clinical Implications and Diagnosis of Vascular Calcification in CKD
4. Treatment Options for Vascular Calcification in CKD
References
- Gao, J.; Zhang, K.; Chen, J.; Wang, M.-H.; Wang, J.; Liu, P.; Huang, H. Roles of aldosterone in vascular calcification: An update. Eur. J. Pharmacol. 2016, 786, 186–193.
- Généreux, P.; Redfors, B.; Witzenbichler, B.; Arsenault, M.-P.; Weisz, G.; Stuckey, T.D.; Rinaldi, M.J.; Neumann, F.-J.; Metzger, D.C.; Henry, T.D.; et al. Two-year outcomes after percutaneous coronary intervention of calcified lesions with drug-eluting stents. Int. J. Cardiol. 2017, 231, 61–67.
- Lee, H.Y.; Park, U.J.; Kim, H.T.; Roh, Y.-N. The Effect of Severe Femoropopliteal Arterial Calcification on the Treatment Outcome of Femoropopliteal Intervention in Patients with Ischemic Tissue Loss. Vasc. Spec. Int. 2020, 36, 96–104.
- Lacolley, P.; Regnault, V.; Laurent, S. Mechanisms of Arterial Stiffening. Arter. Thromb. Vasc. Biol. 2020, 40, 1055–1062.
- Lioufas, N.M.; Pedagogos, E.; Hawley, C.M.; Pascoe, E.M.; Elder, G.J.; Badve, S.V.; Valks, A.; Toussaint, N.D.; on behalf of the IMPROVE-CKD Investigators. Aortic Calcification and Arterial Stiffness Burden in a Chronic Kidney Disease Cohort with High Cardiovascular Risk: Baseline Characteristics of the Impact of Phosphate Reduction on Vascular End-Points in Chronic Kidney Disease Trial. Am. J. Nephrol. 2020, 51, 201–215.
- Jagieła, J.; Bartnicki, P.; Rysz, J. Selected cardiovascular risk factors in early stages of chronic kidney disease. Int. Urol. Nephrol. 2020, 52, 303–314.
- Demer, L.L.; Tintut, Y. Vascular Calcification. Circulation 2008, 117, 2938–2948.
- Chen, N.X.; Moe, S.M. Vascular calcification: Pathophysiology and risk factors. Curr. Hypertens. Rep. 2012, 14, 228–237.
- Hénaut, L.; Chillon, J.-M.; Kamel, S.; Massy, Z.A. Updates on the Mechanisms and the Care of Cardiovascular Calcification in Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 233–250.
- Voelkl, J.; Lang, F.; Eckardt, K.-U.; Amann, K.; Kuro-O, M.; Pasch, A.; Pieske, B.; Alesutan, I. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell. Mol. Life Sci. 2019, 76, 2077–2091.
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular Calcification: An Update on Mechanisms and Challenges in Treatment. Calcif. Tissue Int. 2013, 93, 365–373.
- El Din, U.A.A.S.; Salem, M.M.; Azim, S.E.D.U.A. Vascular calcification: When should we interfere in chronic kidney disease patients and how? World J. Nephrol. 2016, 5, 398–417.
- Nakanishi, T.; Nanami, M.; Kuragano, T. The pathogenesis of CKD complications; Attack of dysregulated iron and phosphate metabolism. Free Radic. Biol. Med. 2020, 157, 55–62.
- Olapoju, S.O.; Adejobi, O.I.; Le Thi, X. Fibroblast growth factor 21; review on its participation in vascular calcification pathology. Vasc. Pharmacol. 2020, 125–126, 106636.
- Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate. Toxins 2020, 12, 58.
- Huang, M.; Zheng, L.; Xu, H.; Tang, D.; Lin, L.; Zhang, J.; Li, C.; Wang, W.; Yuan, Q.; Tao, L.; et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J. Mol. Cell. Cardiol. 2020, 138, 256–268.
- Sarnak, M.J. Cardiovascular complications in chronic kidney disease. Am. J. Kidney Dis. 2003, 41, 11–17.
- Sheng, B.; Zhu, T.; Li, J. End-stage renal disease and pulmonary hypertension. Zhong Nan Da Xue Xue Bao Yi Xue Ban = J. Cent. South Univ. Med. Sci. 2019, 44, 1419–1422.
- Arefin, S.; Buchanan, S.; Hobson, S.; Steinmetz, J.; Alsalhi, S.; Shiels, P.G.; Kublickiene, K.; Stenvinkel, P. Nrf2 in early vascular ageing: Calcification, senescence and therapy. Clin. Chim. Acta 2020, 505, 108–118.
- Lee, S.J.; Lee, I.-K.; Jeon, J.-H. Vascular Calcification—New Insights into Its Mechanism. Int. J. Mol. Sci. 2020, 21, 2685.
- Thompson, B.; Towler, D.A. Arterial calcification and bone physiology: Role of the bone–vascular axis. Nat. Rev. Endocrinol. 2012, 8, 529–543.
- Xu, J.; Shi, G.-P. Vascular wall extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 2106–2119.
- Cucchiari, D.; Torregrosa, J.-V. Calcifilaxis en pacientes con enfermedad renal crónica: Una enfermedad todavía desconcertante y potencialmente mortal. Nefrología 2018, 38, 579–586.
- House, S.J.; Potier, M.; Bisaillon, J.; Singer, H.A.; Trebak, M. The non-excitable smooth muscle: Calcium signaling and phenotypic switching during vascular disease. Pflug. Arch. Eur. J. Physiol. 2008, 456, 769–785.
- Cianciolo, G.; Galassi, A.; Capelli, I.; Schillaci, R.; La Manna, G.; Cozzolino, M. Klotho-FGF23, Cardiovascular Disease, and Vascular Calcification: Black or White? Curr. Vasc. Pharmacol. 2018, 16, 143–156.
- Kapustin, A.N.; Davies, J.D.; Reynolds, J.L.; McNair, R.; Jones, G.T.; Sidibe, A.; Schurgers, L.J.; Skepper, J.N.; Proudfoot, D.; Mayr, M.; et al. Calcium Regulates Key Components of Vascular Smooth Muscle Cell–Derived Matrix Vesicles to Enhance Mineralization. Circ. Res. 2011, 109, e1–e12.
- Houben, E.; Neradova, A.; Schurgers, L.J.; Vervloet, M. The influence of phosphate, calcium and magnesium on matrix Gla-protein and vascular calcification: A systematic review. G. Ital. Nefrol. 2017, 33, 1724–5590.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2009, 113, S1–S130.
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human Vascular Smooth Muscle Cells Undergo Vesicle-Mediated Calcification in Response to Changes in Extracellular Calcium and Phosphate Concentrations: A Potential Mechanism for Accelerated Vascular Calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867.
- Disthabanchong, S. Vascular calcification in chronic kidney disease: Pathogenesis and clinical implication. World J. Nephrol. 2012, 1, 43–53.
- Merjanian, R.; Budoff, M.; Adler, S.; Berman, N.; Mehrotra, R. Coronary artery, aortic wall, and valvular calcification in nondialyzed individuals with type 2 diabetes and renal disease. Kidney Int. 2003, 64, 263–271.
- Lamprea-Montealegre, J.A.; McClelland, R.L.; Astor, B.C.; Matsushita, K.; Shlipak, M.; de Boer, I.H.; Szklo, M. Chronic kidney disease, plasma lipoproteins, and coronary artery calcium incidence: The Multi-Ethnic Study of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 652.
- Kramer, H.; Toto, R.; Peshock, R.; Cooper, R.; Victor, R. Association between Chronic Kidney Disease and Coronary Artery Calcification: The Dallas Heart Study. J. Am. Soc. Nephrol. 2004, 16, 507–513.
- Qunibi, W.Y.; AbouZahr, F.; Mizani, M.R.; Nolan, C.R.; Arya, R.; Hunt, K.J. Cardiovascular calcification in Hispanic Americans (HA) with chronic kidney disease (CKD) due to type 2 diabetes. Kidney Int. 2005, 68, 271–277.
- Russo, D.; Palmiero, G.; De Blasio, A.P.; Balletta, M.M.; Andreucci, V.E. Coronary artery calcification in patients with CRF not undergoing dialysis. Am. J. Kidney Dis. 2004, 44, 1024–1030.
- Chiu, Y.-W.; Adler, S.G.; Budoff, M.J.; Takasu, J.; Ashai, J.; Mehrotra, R. Coronary artery calcification and mortality in diabetic patients with proteinuria. Kidney Int. 2010, 77, 1107–1114.
- Bellasi, A.; Raggi, P. Vascular calcification in patients with kidney disease: Techniques and Technologies to Assess Vascular Calcification. Semin. Dial. 2007, 20, 129–133.
- Uhlig, K. There Is No Practical Utility in Routinely Screening Dialysis Patients for Vascular Calcification. Semin. Dial. 2010, 23, 277–279.
- Beto, J.; Bhatt, N.; Gerbeling, T.; Patel, C.; Drayer, D. Overview of the 2017 KDIGO CKD-MBD Update: Practice Implications for Adult Hemodialysis Patients. J. Ren. Nutr. 2019, 29, 2–15.
- Ix, J.H.; Katz, R.; Kestenbaum, B.; Fried, L.F.; Kramer, H.; Stehman-Breen, C.; Shlipak, M.G. Association of Mild to Moderate Kidney Dysfunction and Coronary Calcification. J. Am. Soc. Nephrol. 2008, 19, 579–585.
- Jun, M.; Lv, J.; Perkovic, V.; Jardine, M.J. Managing cardiovascular risk in people with chronic kidney disease: A review of the evidence from randomized controlled trials. Ther. Adv. Chronic Dis. 2011, 2, 265–278.
- Himmelsbach, A.; Ciliox, C.; Goettsch, C. Cardiovascular Calcification in Chronic Kidney Disease—Therapeutic Opportunities. Toxins 2020, 12, 181.
- Jamal, S.A.; Vandermeer, B.; Raggi, P.; Mendelssohn, D.C.; Chatterley, T.; Dorgan, M.; Lok, C.E.; Fitchett, D.; Tsuyuki, R.T. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: An updated systematic review and meta-analysis. Lancet 2013, 382, 1268–1277.
- Block, G.A.; Bushinsky, D.A.; Cheng, S.; Cunningham, J.; Dehmel, B.; Drueke, T.B.; Ketteler, M.; KewalRamani, R.; Martin, K.J.; Moe, S.M.; et al. Effect of Etelcalcetide vs. Cinacalcet on Serum Parathyroid Hormone in Patients Receiving Hemodialysis with Secondary Hyperparathyroidism. JAMA 2017, 317, 156–164.
- Chitalia, N.; Recio-Mayoral, A.; Kaski, J.C.; Banerjee, D. Vitamin D deficiency and endothelial dysfunction in non-dialysis chronic kidney disease patients. Atherosclerosis 2012, 220, 265–268.
- Perazella, M.A.; Markowitz, G.S. Bisphosphonate nephrotoxicity. Kidney Int. 2008, 74, 1385–1393.
- Bergner, R.; Diel, I.J.; Henrich, D.; Hoffmann, M.; Uppenkamp, M. Differences in Nephrotoxicity of Intravenous Bisphosphonates for the Treatment of Malignancy Related Bone Disease. Oncol. Res. Treat. 2006, 29, 534–540.
- Louvet, L.; Büchel, J.; Steppan, S.; Passlick-Deetjen, J.; Massy, Z.A. Magnesium prevents phosphate-induced calcification in human aortic vascular smooth muscle cells. Nephrol. Dial. Transplant. 2012, 28, 869–878.
- Sharples, E.J.; Pereira, D.; Summers, S.; Cunningham, J.; Rubens, M.; Goldsmith, D.; Yaqoob, M.M. Coronary artery calcification measured with electron-beam computerized tomography correlates poorly with coronary artery angiography in dialysis patients. Am. J. Kidney Dis. 2004, 43, 313–319.
- Maniscalco, B.S.; Taylor, K.A. Calcification in coronary artery disease can be reversed by EDTA–tetracycline long-term chemotherapy. Pathophysiology 2004, 11, 95–101.
- Krieger, N.S.; Frick, K.K.; Bushinsky, D.A. Mechanism of acid-induced bone resorption. Curr. Opin. Nephrol. Hypertens. 2004, 13, 423–436.
- Mendoza, F.J.; Lopez, I.; Montes de Oca, A.; Perez, J.; Rodriguez, M.; Aguilera-Tejero, E. Metabolicacidosis inhibits soft tissue calcification in uremic rats. Kidney Int. 2008, 73, 407–414.
- Simpson, C.L.; Lindley, S.; Eisenberg, C.; Basalyga, D.M.; Starcher, B.C.; Simionescu, D.T.; Vyavahare, N.R. Toward cell therapy for vascular calcification: Osteoclast-mediated demineralization of calcifiedelastin. Cardiovasc. Pathol. 2007, 16, 29–37.


